
What is Hi-C
Hi-C (Chromosome conformation capture followed by high-throughput sequencing) is a method used to study the three-dimensional structure of the genome. The Hi-C technique originated from Chromosome Conformation Capture (3C) technology and involves the use of high-throughput sequencing and bioinformatics analysis to investigate the spatial relationships of chromatin DNA throughout the entire genome. This enables the acquisition of high-resolution three-dimensional structural information of chromatin.
Hi-C principle
The Chromosome Conformation Capture (3C) technology, which is part of the Hi-C method, involves cross-linking chromatin in the cell nucleus using formaldehyde, digesting the chromatin-protein complexes with an excess of restriction enzymes, and ligating the digested fragments under conditions of low DNA concentration and high ligase concentration. The cross-linked proteins are then digested to release bound proteins. Ordinary PCR and quantitative PCR using primers designed for potentially interacting fragments are used to determine whether there is any interaction. The 3C technique assumes that DNA fragments that interact physically have the highest ligation frequency. Gene locus-specific PCR is used to detect physical contacts between DNA fragments in the genome. Ultimately, the presence of interactions is determined based on the abundance of PCR products.
In essence, chromatin is enveloped within a 3D structure, and the closer the sequences are on the same chromosome, the closer they are in space. Therefore, the goal is to identify which contigs are closer in space to infer their proximity on the same chromosome.
Hi-C Workflow
Formaldehyde cross-linking:
The sample is fixed using formaldehyde, which cross-links proteins with DNA and DNA with DNA, preserving their interaction relationships and maintaining the 3D structure within the cell. Typically, live samples are treated with 1-3% formaldehyde at room temperature for 10-30 minutes. However, this step may reduce the efficiency of restriction enzyme digestion of DNA sequences and requires strict control.
Enzymatic digestion:
DNA is digested using restriction enzymes, resulting in fragments with sticky ends on both sides of the cross-linking points. The size of the resulting fragments affects the sequencing resolution. Two types of enzymes are commonly used: 6-base pair (bp) restriction enzymes and 4-bp restriction enzymes. The latter provides higher resolution. Enzymes such as EcoR1 or HindIII are used to cut the genome every 4000 bp, generating approximately one million fragments in the human genome.
End repair:
The resulting fragments have either blunt ends or sticky ends, which are then repaired to generate blunt ends. The end repair mechanism is used to introduce biotin-labeled nucleotides for subsequent DNA purification and capture.
Ligation:
The repaired DNA fragments are ligated using T4 DNA ligase to create circles, bringing together DNA fragments that interact with each other. The protein connecting the DNA fragments is then digested, resulting in cross-linked fragments.
DNA purification and capture:
The DNA is de-cross-linked, purified, and fragmented into 300 bp - 700 bp fragments. These fragments, which contain DNA segments with interacting relationships, are captured using biotinylated probes to construct libraries. The fragments are further fragmented using techniques such as sonication.
Sequencing:
The captured DNA, bound to magnetic beads, is used to create libraries for sequencing, which is performed using high-throughput sequencing technologies.
Hi-C Applications
Studying changes in the interaction patterns of specific chromosomal regions under different conditions.
Performing joint analysis with other data, such as ChIP-Seq, to elucidate the association between epigenetic modifications, transcription factors, and chromosomal structure.
Integrating expression profiling data, such as RNA-Seq, to investigate the impact of chromosomal 3D structure on transcriptional activity.
Constructing chromosome folding models.
Advantages of Hi-C Technique
Utilizes advanced in situ Hi-C technology, effectively reducing experimental noise and biases associated with traditional Hi-C methods.
Achieves a high data yield with a high unique mapping rate (up to 80% of uniquely mapped data).
Provides high-resolution information about chromosomal structure.
Hi-C Data Analysis
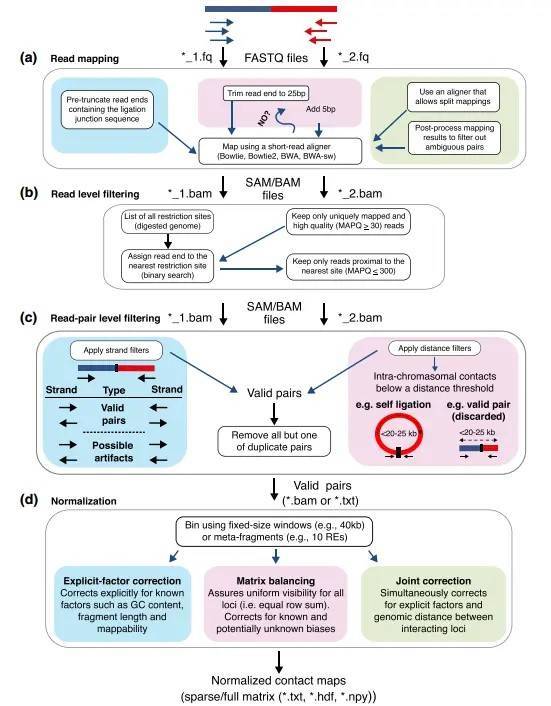
Analysis includes standard analysis and advanced analysis. The construction of chromosomal domains using genome-wide interaction information allows for obtaining high-resolution information about chromatin's three-dimensional structure and the identification of regulatory DNA elements involved in gene regulation. Furthermore, the integration of transcriptomic data, resequencing, and ChIP-Seq data allows for a comprehensive analysis of biological questions, including in-depth exploration of various aspects such as base changes, gene expression, protein modifications, and chromosomal three-dimensional conformation analysis.
| Analysis Content | Description |
|---|---|
| Construction of Chromosomal Haplotypes | Building haplotypes for chromosomes |
| 3D Structure Reconstruction and Regulatory Element Development | Reconstructing the three-dimensional structure of chromatin and developing regulatory elements based on it |
| Sequencing Quality Assessment | Evaluating the quality of sequencing data |
| Hi-C Data Quality Control and Alignment Statistics | Performing quality control and alignment statistics for Hi-C data |
| Insert Fragment Statistics | Counting the number of inserted fragments in Hi-C experiments |
| Alignment to NT Library | Aligning the data to the NT library |
| Alignment to Reference Genome | Aligning and filtering the data against a reference genome |
| SNP Detection and Annotation | Detecting and annotating single nucleotide polymorphisms (SNPs) |
| Construction of Interaction Maps | Creating interaction maps based on Hi-C data |
| InDel Detection and Annotation | Detecting and annotating insertions/deletions (InDels) |
| TAD Analysis | Analyzing Topologically Associating Domains (TADs) in the genome |
| Genome Heterozygosity Statistics | Statistical analysis of genome heterozygosity |
| 3D Structure Reconstruction | Reconstructing the three-dimensional structure of chromatin through data analysis |
| Construction of Chromosomal Haplotypes | Building haplotypes for chromosomal spans |
| Development of Regulatory Elements | Discovering and studying regulatory elements for gene expression |
Comparative Hi-C Reveals that CTCF Underlies Evolution of Chromosomal Domain Architecture
Journal: Cell Reports
Publicated: 2015
Background: CTCF (CCCTC-binding factor) is a multifunctional transcription factor widely present in eukaryotes. It is also known as an 11-zinc finger protein and is closely associated with the activity of insulators, which are regulatory elements in the eukaryotic genome that function to block or enhance the nearby regulatory elements, exerting effects on the promoters of the genes they define. The human genome contains nearly 15,000 CTCF insulator sites, indicating the widespread functional role of CTCF in gene regulation. In March 2015, Vietri-Rudan et al. reported the use of Hi-C technology to uncover the important role of CTCF in promoting genome structural changes. This study compared CTCF binding sites in four mammalian species and revealed a direct connection between insulator site divergence and chromatin structural domains. The findings were published in Cell Reports (IF=8.358), providing new insights and methods for the study of transcription factors.
Results
- Mechanism of CTCF function:
CTCF and cohesin proteins exert their effects on genes by forming stable chromatin loops. These CTCF/cohesin-anchored loop structures are widely distributed throughout the genome, encompassing not only loop structures that define topological domains but also loop structures within these domains. - Sequence variation drives CTCF binding variation:
CTCF recognizes and binds to specific DNA sequences. ChIP-seq data for CTCF in mice, dogs, and macaques indicate that chromatin regions with strong CTCF binding have relatively conserved sequences. Sequence variations can alter CTCF's ability to bind, thereby affecting CTCF binding. These results suggest that sequence variations drive variations in CTCF binding. - Evolutionary dynamics of CTCF and its correlation with chromosomal topological domains:
Hi-C data from mouse liver cells show that conserved CTCF sites tend to be located at the boundaries of chromosomal topological domains, while species-specific CTCF sites tend to be located within these domains. This indicates a correlation between the evolutionary dynamics of CTCF and chromosomal topological domains. - Comparison of chromosomal topological domains using Hi-C data:
Hi-C data from mouse, macaque, dog, and rabbit indicate similarities in their topological domains, with the differences primarily found within the domains. Comparing the chromosomal topological structures of dogs and mice reveals that conserved domains are slightly smaller than non-conserved domains. - CTCF binding variation drives chromosomal structural variations:
CTCF binding influences the internal structure of chromosomal domains, but has less impact on larger-scale structural domains. Partially conserved CTCF sites (e.g., present in mice and dogs but absent in macaques) lead to reduced insulator effects when they are absent. This suggests that CTCF binding variation drives chromosomal structural variations. - Connectivity between different topological domains:
Within the conserved CTCF sites in mice and dogs, 94% show conserved orientation, indicating a directional aspect of CTCF sites. Additionally, there is connectivity between different topological domains.
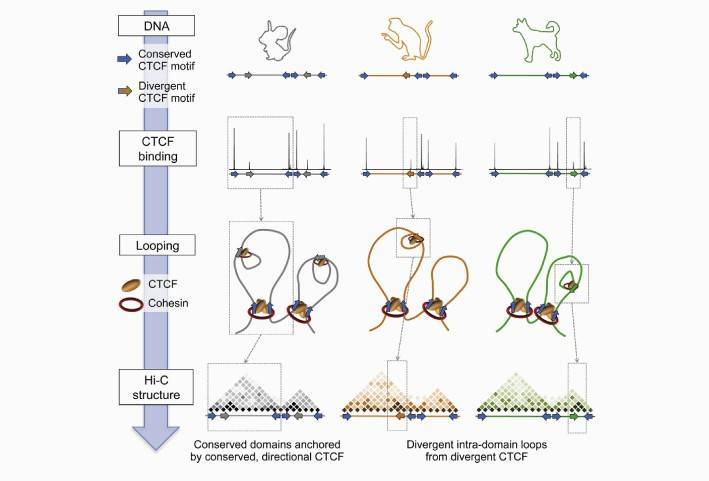
Conclusions
CTCF recognizes and binds to specific DNA sequences. ChIP-seq data for CTCF in mice, dogs, and macaques indicate that chromatin regions with strong CTCF binding have relatively conserved sequences. CTCF binding influences the internal structure of chromosomal domains, while larger-scale structural domains are not affected by CTCF binding.