
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Nanopore sequencing technology holds several prominent advantages, including, in broad strokes, long read lengths, high output, portability, real-time operation, ease of use, and direct reading capabilities. A more detailed explanation of these advantages can be found in the article "Why Choose Nanopore Sequencing?". These characteristics render this technology immensely valuable within the realm of genomic or transcriptomic analyses. This document aims to provide a list of typical use cases of Nanopore sequencing technology in actual applications as references for readers.
Nanopore sequencing finds applications in diverse research domains: genome assembly (e.g., Mb-level long-read sequencing), direct detection of base modifications (e.g., DNA/RNA sequencing), chromosome-level targeted sequencing, and rapid real-time sequencing of metagenomes (pathogenic microorganism detection).
Service you may intersted in
Resource
One of the most distinctive features of nanopore sequencing technology is its capability for long-read sequencing, a crucial advantage in large genome assembly. In the past, genome assembly based on short fragments posed significant challenges, especially for organisms with characteristics such as polyploidy, extensive repeats, and high heterozygosity. For certain plants like Paris japonica with a genome size of 152 Gb and the decaploid Fragaria turupensis, short-read assembly was inadequate. Despite the introduction of techniques like large-insert libraries, BAC libraries, optical mapping, Hi-C, and bionano, the fundamental challenges of large genome assembly persisted. Nanopore sequencing, however, proves instrumental in addressing these challenges and significantly enhances genome completeness.
Thanks to the real-time, fast, and compact characteristics of nanopore sequencing technology, it enables direct sequencing, facilitating immediate acquisition of sequence information for species classification and identification, thus enabling rapid microbial recognition. With the enhancement of sequencing throughput, real-time monitoring has been widely applied to pathogens with larger genomes, from viruses of just a few kilobases (Kb), through to bacteria with a few megabases (Mb), and even human fungal pathogens with genomes larger than 10 Mb, all of which can be monitored onsite and in real-time.
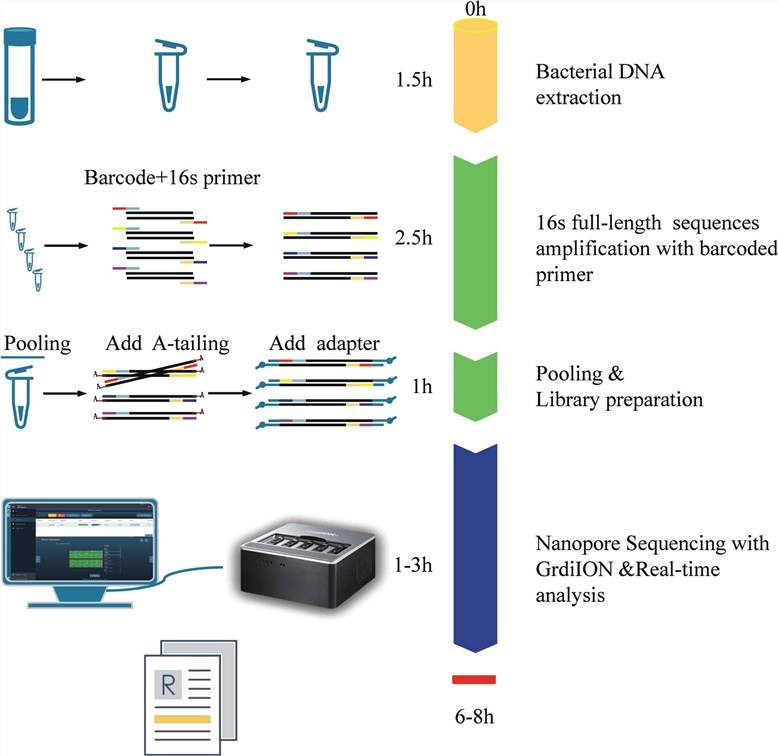 The process and schematic representation of 16S rDNA nanopore sequencing. The turnaround time was approximately 6~8 h from sample collection to results. (Yinghu Chen et al., 2023)
The process and schematic representation of 16S rDNA nanopore sequencing. The turnaround time was approximately 6~8 h from sample collection to results. (Yinghu Chen et al., 2023)
The emergence of antibiotic resistance through genetic mutations in pathogens poses a significant challenge to clinical treatment. Currently, Antimicrobial Susceptibility Testing (AST) is the primary method for diagnosing microbial drug resistance. However, standard AST methods have limitations. Nanopore sequencing technology excels in detecting mutations in targeted drug-resistant genes, offering real-time guidance for clinical treatment and serving as a reference for patient prognosis assessment. The high-throughput, high-output, and efficient sequencing platform provides crucial clinical value for achieving real-time monitoring.
Historically, transcriptome analysis involved the sequencing of mRNA after fragmentation and reverse transcription to cDNA. In this process, PCR may introduce amplification bias, leading to the identification of false-positive differentially expressed genes. Despite the current use of single-cell methods with molecular barcodes to reduce PCR bias, obtaining and analyzing full-length transcripts remains a challenge. Due to the splice variations prevalent in eukaryotes, short-read sequencing still struggles with accurate identification. Utilizing nanopore long-read sequencing, however, enables the precise identification of multiple isoforms for each gene, offering a simplified and accurate approach.
Learn more: Full-length Transcript Sequencing: A Comparison Between PacBio Iso-Seq and Nanopore Direct RNA-Seq
Nanopore sequencing technology stands out as the sole method capable of directly sequencing RNA chains without the need for reverse transcription and amplification. This approach exhibits no sequencing preference and accurately detects methylation sites on RNA molecules, such as m6A and m5C modifications. Additionally, nanopore sequencing provides a relatively precise estimation of Poly(A) tail lengths, contributing to the revelation of authentic RNA features.
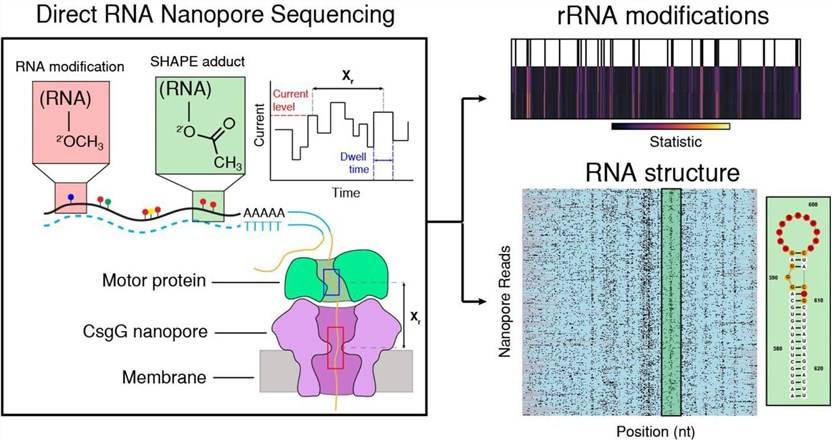 Direct detection of RNA modifications and structure using single molecule nanopore sequencing. (William Stephenson,. et al., Cell Genomics 2022)
Direct detection of RNA modifications and structure using single molecule nanopore sequencing. (William Stephenson,. et al., Cell Genomics 2022)
Genomic variations encompass Single Nucleotide Variants (SNVs), Insertions and Deletions (Indels), and Structural Variants (SVs). Whole Exome Sequencing (WES), based on short-read sequencing, faces limitations in detecting structural variations with a detection rate below 50%. This is attributed to the instability of short-read sequencing in identifying repetitive regions and its challenge in covering high GC-content areas. However, current research confirms the significance of characterizing these repetitive regions and structural variations in relation to human health and disease conditions such as aging, trinucleotide repeat disorders, autism, epilepsy, and cancer. Therefore, routine identification and characterization of these variations hold paramount importance. Nanopore sequencing technology, with its advantages of ultra-long reads (currently up to 2.4 MB), lack of GC bias, and direct detection of methylation, surpasses the challenges faced by short-read sequencing in identifying genetic SVs by spanning across repetitive sequences.
Haplotype analysis, also known as phasing, fundamentally addresses allele information in diploid or polyploid genomes, distinguishing their parental origins. This ensures that information from the same parent is orderly arranged on the respective chromosomes. Clearly, the increase in sequencing length holds positive implications for haplotype analysis (especially when dealing with complete chromosomes, allowing for direct alignment). According to literature, the application of ultra-long reads not only doubles the continuity of assemblies (NG50 ~ 6.4 MB) but also significantly enhances the phasing effectiveness of allelic genes. For instance, achieving haplotyping within a single 16 MB contig, especially for the 4 MB Major Histocompatibility Complex (MHC), becomes a feasible prospect.
Given the relatively compact size of bacterial genomes, typically not exceeding 10 Mb, and their predominantly single-chromosome structure with minimal repetitive sequences, the long-read capability of nanopore technology enables the one-step assembly of entire genomes within a few minutes (depending on the specific sample and hardware configurations).
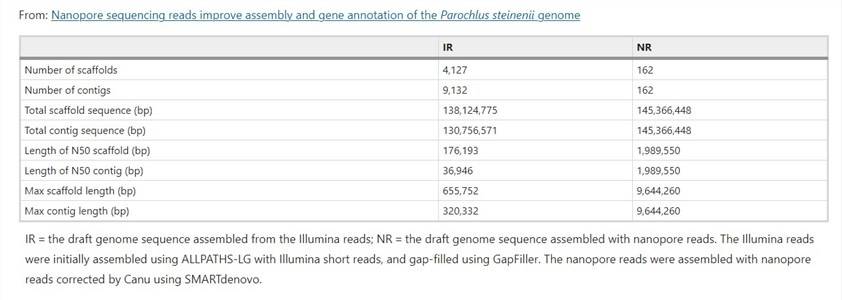 Nanopore sequencing reads improve assembly and gene annotation of the Parochlus steinenii genome (Shin, S.C. et al., 2019)
Nanopore sequencing reads improve assembly and gene annotation of the Parochlus steinenii genome (Shin, S.C. et al., 2019)
While long read sequencing platforms, represented by Nanopore sequencing technology, may exhibit lower accuracy compared to second-generation platforms, their emergence as a novel technology, diverse array of reagents, and lack of standardized analysis methods contribute to this. Nevertheless, these platforms demonstrate commendable performance in microbial identification, pathogen detection, oncological research, and the detection of genetic diseases. Despite their current limitations, there exists significant potential for advancement, offering new perspectives and techniques for relevant scientific disciplines. Nanopore sequencing is poised to unfold greater research value across various fields.
References:


