Single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics are advanced techniques that exhibit unique advantages in elucidating cellular heterogeneity and tissue architecture. These methodologies have garnered significant attention across various research domains.
Single-Cell RNA Sequencing
Single-cell RNA sequencing (scRNA-seq) represents a novel approach to high-throughput sequencing of mRNA at the single-cell level. This technique involves the efficient amplification of minimal amounts of mRNA extracted from isolated individual cells, followed by high-throughput sequencing. Applications of single-cell sequencing span several fields, including developmental biology, oncology, neuroscience, immunology, the study of disease mechanisms, and the construction of cellular atlases.
Rationale for Single-Cell Transcriptome Analysis
Cells do not operate in isolation; rather, they collaborate to fulfill biological functions. As the adage suggests, "no two leaves are exactly alike," each cell possesses unique characteristics and performs distinct roles within its environment.
Traditional transcriptomic methods, such as bulk RNA sequencing (Bulk RNA-seq), necessitate the grinding and extraction of mixed total RNA from organisms, tissues, or cell populations for sequencing. Consequently, the resulting data represent averaged transcriptomic profiles from diverse cellular constituents. This approach tends to overlook intercellular differences and obscures cellular heterogeneity. In fact, Bulk RNA-seq often describes a hypothesized state, which may not accurately reflect the actual status of individual cells.
In contrast, scRNA-seq technology can reveal the heterogeneity and complexity of RNA transcripts within single cells, as well as delineate the composition of various cell types and functions in highly organized tissues, organs, and organisms. This technological advancement has profoundly transformed the field of transcriptomics.
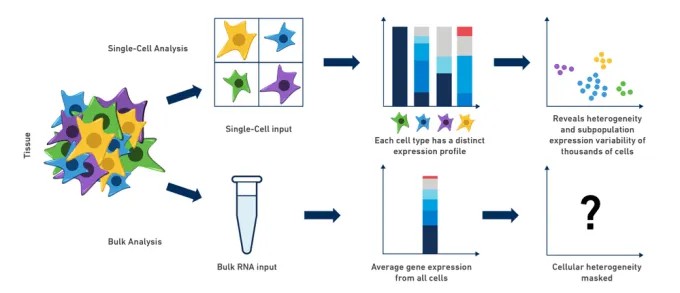 Figure 1: Comparison of scRNA-seq and Bulk RNA-seq
Figure 1: Comparison of scRNA-seq and Bulk RNA-seq
Technical Advantages
Revelation of Cellular Heterogeneity and High-Resolution Analysis: This technique facilitates the identification of cellular subpopulations and rare cell types, providing comprehensive insights into cellular functions and states.
Investigation of Dynamic Processes: scRNA-seq is suitable for exploring the evolution of gene expression during dynamic processes such as cell differentiation, proliferation, and tumorigenesis.
Unbiased Cell Clustering: The method allows for more precise classification of cells, particularly within the fields of immunology, oncology, and genetics.
Implementation of Single-Cell Transcriptomics
The primary distinction between the operational workflow of scRNA-seq and traditional Bulk RNA-seq lies in the requirement to isolate individual cells. A typical scRNA-seq process consists of the following steps: sample collection, single-cell isolation, cell lysis, reverse transcription, cDNA amplification, library construction, sequencing, and data analysis. Current mainstream scRNA-seq technology platforms include:
Microfluidic Platforms: Examples include 10x Genomics, MobiNova, and DNBelab C4, which utilize microfluidic chips and the principle of immiscibility between oil and water for single-cell isolation.
Microplate Platforms: For instance, the BD Rhapsody platform employs gravity to facilitate the deposition of cells into micro-wells for separation.
Spatial Transcriptomics
Spatial transcriptomics enables the detection of the entire transcriptome expression within cells while preserving their spatial location within the tissue. This approach allows for a more in-depth exploration of spatial heterogeneity. Its applications encompass the identification of rare cell types, studies of growth and development, exploration of disease mechanisms, and analysis of the tumor microenvironment.
In single-cell sequencing, sample cells may be lost during tissue dissociation and microfluidic processing, potentially leading to distortion of information. Moreover, single-cell sequencing often results in the loss of spatial positional data for the cells. Spatial transcriptomics addresses the deficiency of spatial information that is inherent in single-cell transcriptomics.
Technical Advantages
Retention of Spatial Information: This methodology preserves the spatial positions of mRNA within tissue sections, revealing intercellular interactions and organizational structure, making it particularly applicable to studies involving morphologically distinct tissues such as the brain, embryos, and olfactory bulbs.
Comprehensive Transcriptomic Analysis: It provides an extensive overview of mRNA expression at the cellular level, enhancing the understanding of cellular activity within complex tissues. In oncology research, this technique can uncover gene expression gradients and heterogeneity within tumor regions.
Implementation of Spatial Transcriptomics
Currently, various methods exist for implementing spatial transcriptomics, including imaging techniques based on in situ hybridization and those relying on transcriptomic sequencing. Among these, the high-throughput transcriptomic sequencing platform, 10x Visium, is one of the more established commercial technologies.
The 10x Visium platform utilizes microarray technology to deposit ordered oligonucleotide arrays onto glass slides. Subsequently, thin tissue sections are placed over the array, and permeabilization allows the diffusion of cellular RNA to oligonucleotides containing barcodes. In situ reverse transcription generates cDNA with spatial indices, which is then subjected to high-throughput sequencing.
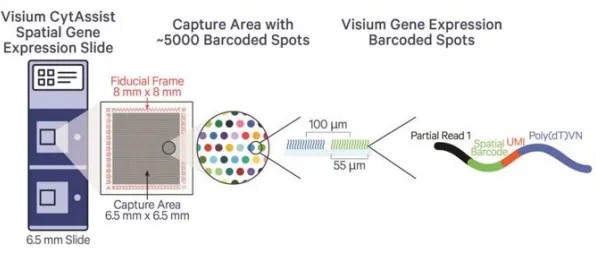 Figure 2: Principles of 10x Visium Spatial Transcriptomics Sequencing
Figure 2: Principles of 10x Visium Spatial Transcriptomics Sequencing
You may interested in
Learn More
Integrated Analysis of Single-Cell and Spatial Transcriptomics Data
In recent years, emerging spatial transcriptomics technologies have enabled the reconstruction of tissue architecture and cellular spatial positioning. However, the resolution of these technologies often falls short of achieving single-cell precision. Therefore, the integration of spatial transcriptomics with single-cell transcriptomics can create high-resolution spatial single-cell maps, yielding a synergistic effect that exceeds the individual applications of each technique. The following sections will explore the conceptual frameworks for the association analysis of these two methodologies, supplemented by case studies to illustrate their application.
Analysis Methods and Principles
Two primary analytical approaches for integrating single-cell transcriptomic data with spatial transcriptomic data are deconvolution and mapping. Fundamentally, these methods involve the integration of single-cell maps, as provided by single-cell transcriptomics, with spatial maps derived from spatial transcriptomics, thereby constructing a spatial single-cell atlas. The subsequent sections will emphasize the analytical strategies and methodologies employed in this integration process.
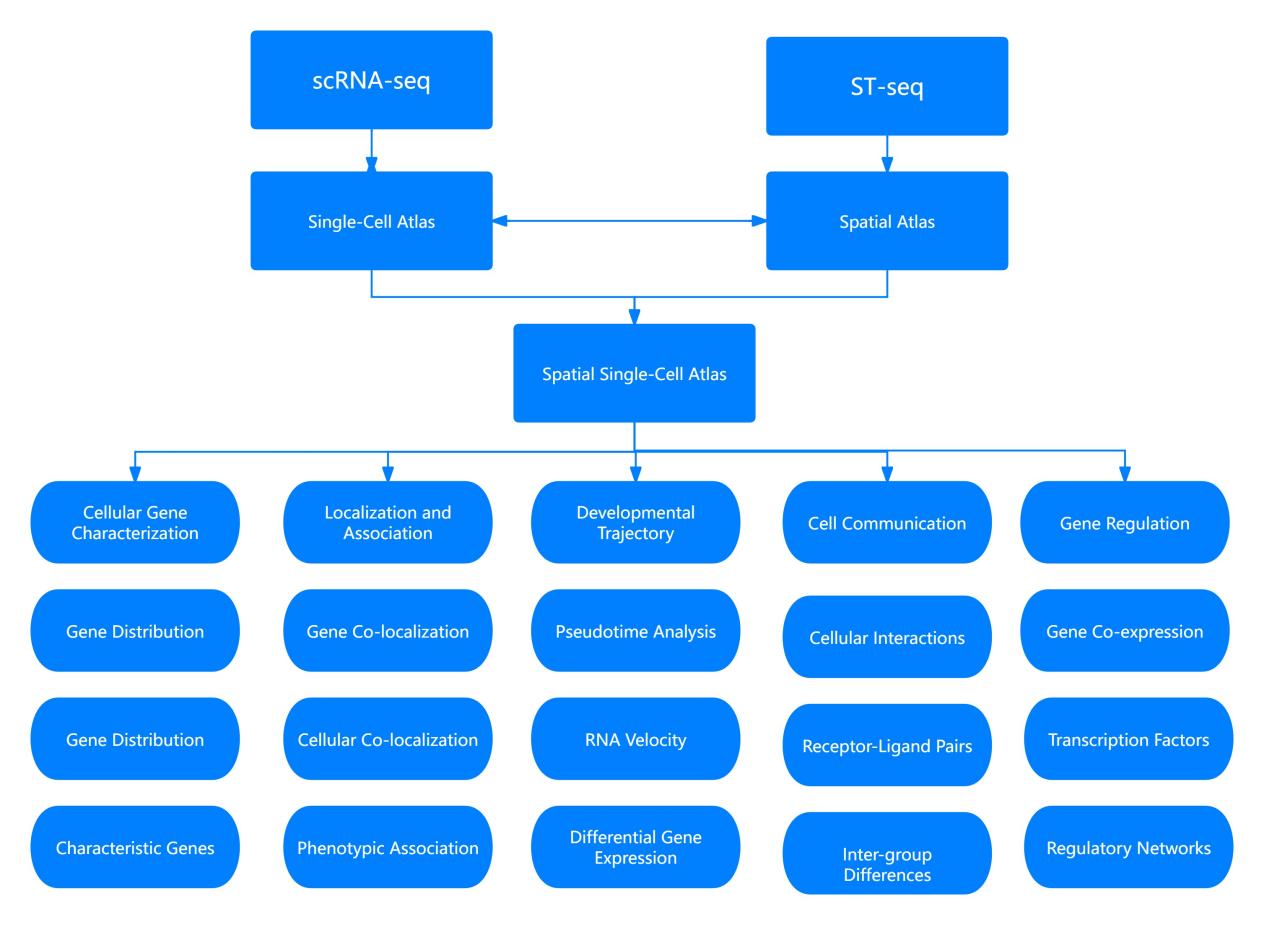 Figure 3: Strategic Approaches to Integrative Analysis of Single-Cell and Spatial Transcriptomics
Figure 3: Strategic Approaches to Integrative Analysis of Single-Cell and Spatial Transcriptomics
Deconvolution
Deconvolution refers to the process of separating mixed transcriptomic signals obtained from spatially resolved samples into their constituent cell types. By leveraging the single-cell transcriptomic profiles, this method allows for the estimation of the abundance of different cell types within specific spatial regions of a tissue sample.
Mapping
Mapping entails aligning single-cell transcriptomic data onto the spatial transcriptomic framework. This method enables the visualization of single-cell expression profiles within the context of tissue architecture, facilitating a deeper understanding of cellular interactions and functional states in situ.
The integration of these methodologies presents a powerful strategy for elucidating the complexities of tissue organization and cellular behavior. Through careful application of deconvolution and mapping techniques, researchers can derive invaluable insights into the cellular composition and functional dynamics of various biological systems.
Cellular Gene Characteristics
The construction of spatial single-cell maps enables the investigation of the distribution patterns of target cell subpopulations, as well as the spatial and subpopulation-specific distribution of target genes. Subsequently, differential gene analysis and enrichment analysis may be conducted to identify genes with the highest upregulation as characteristic genes of the subpopulation, thereby elucidating their distribution across spatial and diverse cell subpopulations.
Localization and Correlation
Further analysis can explore the co-localization of genes and cells, revealing receptor-ligand interactions and transcriptional regulatory relationships, as well as intercellular interactions. Additionally, spatial regions, cells, and genes may be correlated with tissue phenotype data, allowing for the investigation of the relationship between spatial feature distributions and phenotypic functions.
Developmental Trajectories
Subsequently, spatial pseudo-time analysis can be employed to construct the spatial pseudo-time differentiation trajectories of cells. By combining differential gene analysis, spatially relevant developmental feature genes and differentiation-driving genes of subpopulations can be identified, thereby illuminating the regulatory mechanisms of cellular developmental differentiation. Furthermore, RNA velocity analysis can assist in inferring the directional dynamics of cell differentiation, serving to validate the results obtained from pseudo-time analyses.
Cell Communication
Moreover, insights into the interaction relationships among cell subpopulations can be derived from spatial positioning data and receptor-ligand interactions. After identifying key interacting cell pairs, further investigation into their receptor gene pairs and molecular regulatory mechanisms can be pursued, as well as analyses of inter-group differences to assess the impact of experimental treatments on cell communication.
Gene Regulation
Finally, the integration of transcription factor analysis and network analysis may facilitate the construction of transcriptional regulatory networks. This approach allows for the identification of key genes and transcription factors, as well as the examination of their spatial co-expression, thereby revealing the regulatory mechanisms of critical pathways and their associations with phenotypes.
Application Cases
The subsequent sections will illustrate how the aforementioned research strategies are applied through two specific case studies.
Case Study 1
In a 2023 article published in Cell (Impact Factor = 45.5), the authors performed single-cell transcriptomics and spatial transcriptomics on various brain regions of humans at 6-23 gestational weeks (GW). This study constructed a spatiotemporal atlas of the human brain, revealing the spatial distribution patterns of different cell types (see Figure 2AB). The authors further explored the heterogeneity of human neural progenitor cells (NPCs), classifying them into five subpopulations. Among these, radial glial cells (RG) emerged as the earliest population, exhibiting significant regional heterogeneity. Additionally, the transcription factor TFAP2C was specifically expressed in RG cells and was found to determine their fate. This study effectively utilized the first research strategy to investigate the spatial distribution characteristics of cell subpopulations within the human brain.
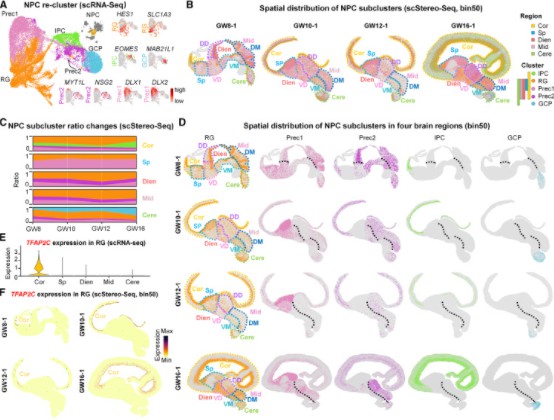 Figure 4: Spatiotemporal Transcriptomic Atlas of Human Brain Development
Figure 4: Spatiotemporal Transcriptomic Atlas of Human Brain Development
Subsequently, the authors employed pseudo-time analysis to reconstruct the developmental trajectories of specific neuronal subpopulations from five distinct brain regions. It was observed that cortical radial glial cells (vRG and oRG) differentiate into brain-specific intermediate progenitor cells (IPC) and precursor cells (Prec2), ultimately developing into cortical glutamatergic neurons (Glu1 and Glu4). Furthermore, the Sp-RG in the ganglionic eminence (GE) progresses toward GABAergic neurons in the cortex.
Additionally, the radial glial cells in the diencephalon (Dien RG-1) are inclined to differentiate into LHX6+ GABAergic neurons, whereas Dien RG-2 is directed toward developing glutamatergic neurons. Spatial visualization results demonstrate that Dien RG-1 and LHX6+ GABAergic neurons are primarily located in the ventral region of the diencephalon, while Dien RG-2 and diencephalic glutamatergic neurons are predominantly situated in the dorsal region. This spatial distribution further corroborates the findings derived from single-cell data analysis.
In summary, the third research strategy was utilized to explore the development of specific neuronal subtypes across different brain regions through spatial pseudo-time analysis.
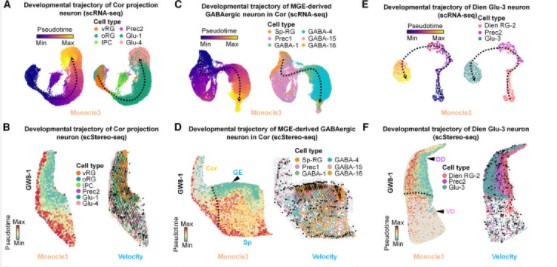 Figure 5: Analysis of Pseudo-Time Spatial Distribution of Target Cell Subpopulations
Figure 5: Analysis of Pseudo-Time Spatial Distribution of Target Cell Subpopulations
Finally, the authors conducted a cell communication analysis to investigate the interactions between glial cells and GABAergic neurons. It was observed that ligand-associated genes NCAM1 and NRXN1 exhibited high expression levels in early oligodendrocyte precursor cells (OPC-1) and GABA-7, while receptor-associated genes L1CAM and NLGN1 were enriched in GABA-7 and early OPC-1, respectively (see Figure 4A). These interactions were predominantly identified within the ventral diencephalon (VD).
Additionally, the analysis revealed interactions between glial cells and neurons from other regions. Early OPC-2 was found to be localized at the boundary between the cortex (Cor) and subpallium (Sp), while glutamatergic neurons Glu1 and Glu4 secreted the ligand NRG1, which attracted OPCs to migrate toward the cortex.
In summary, the fourth research strategy was employed to elucidate the interactions between glial cells and neurons, as well as to characterize the spatial distribution preferences of glial cells during the developmental process.
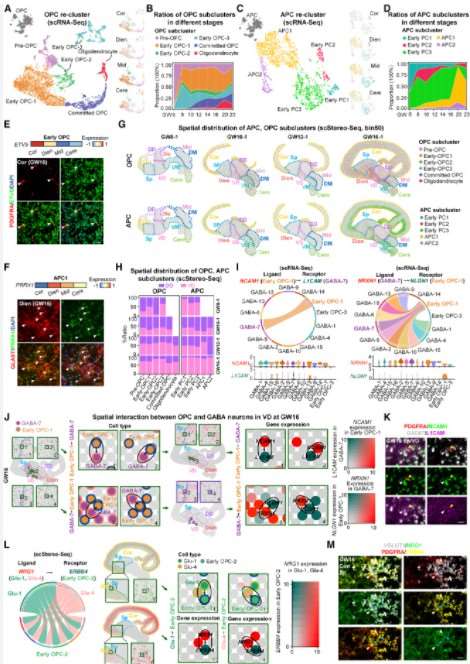 Figure 6: Spatial Distribution and Interactions of Glial Cells
Figure 6: Spatial Distribution and Interactions of Glial Cells
Case Study Two
In a study published in Cell Discovery (Impact Factor = 33.5) in 2024, the authors conducted a spatial single-cell multi-omics analysis of the human cerebellum. The investigation identified GABAergic lineages, such as interneurons and Purkinje cells (PKCs), as well as excitatory lineages, including unipolar brush cells (UBCs), granule cell (GC) progenitors, and post-mitotic GCs.
The analysis revealed that the abundance of PKCs peaked at gestational week 13 (GW13) and subsequently exhibited a sharp decline. Spatial transcriptomic data indicated a multilayered, circumferential distribution of different cell types at GW13, corresponding to the outer external granule layer (oEGL), inner external granule layer (iEGL), intermediate zone (IZ), and ventricular zone (VZ). At gestational week 16 (GW16), notable separations of the rhombic lip (RL), external granule layer (EGL), and Purkinje cell layer (PCL) were also detected .
In summary, this study utilized the first research strategy to investigate the spatial distribution characteristics of cerebellar cell subpopulations.
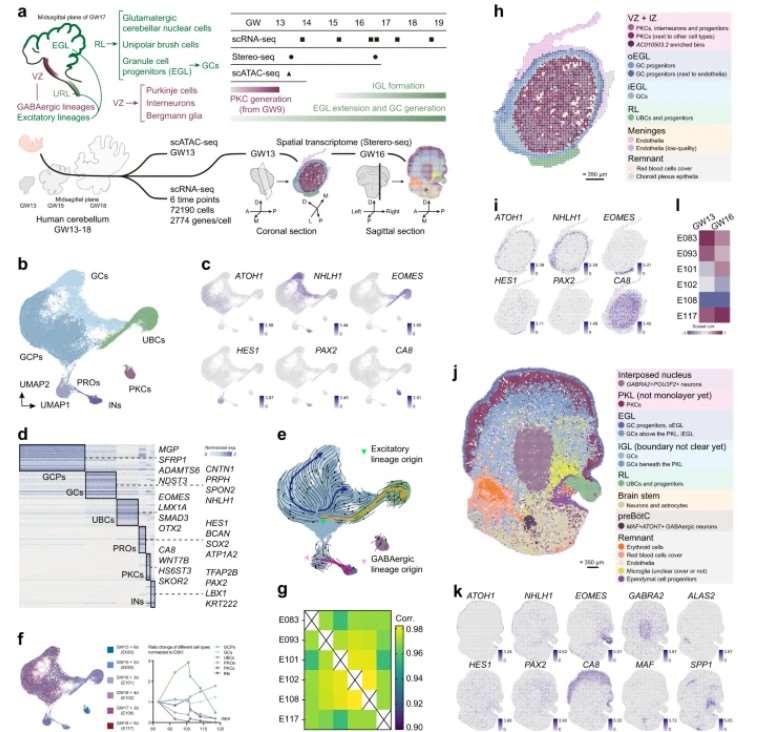 Figure 7: Temporal and Spatial Transcriptomic Map of Human Cerebellar Development
Figure 7: Temporal and Spatial Transcriptomic Map of Human Cerebellar Development
In the subsequent analysis, the authors employed pseudo-time analysis to reconstruct the developmental trajectory of granule cell (GC) lineages. The granule cell progenitors (GCPs) were categorized into two distinct populations: ATOH1+ GCPs (AT+GCPs) and NEUROD1+ GCPs (ND+GCPs). At gestational week 13 (GW13), the ND+GCPs were observed to be positioned beneath the AT+GCPs, exhibiting a distinct inner-outer separation in their distribution.
Further analysis involved selecting transcription factors that were enriched in progenitor or post-mitotic cells for gene regulatory network (GRN) analysis. This approach aimed to identify key genes that may regulate cellular state transitions during GC differentiation. It was revealed that, in addition to ATOH1, transcription factors such as YBX3 and MEF2C may also play roles in GC development.
In summary, this study utilized the third and fifth research strategies to investigate the differentiation and regulatory mechanisms of the GC lineage.
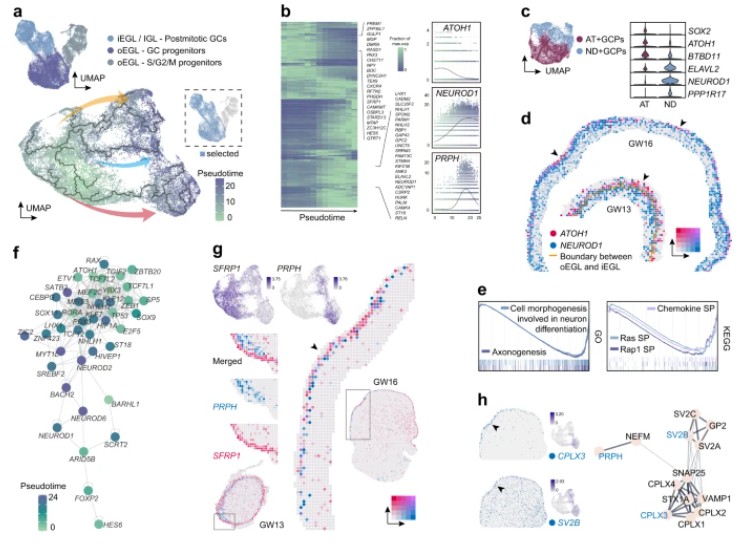 Figure 8: Spatial Distribution of Granule Cell Lineages and Differentiation Regulatory Network
Figure 8: Spatial Distribution of Granule Cell Lineages and Differentiation Regulatory Network
Summary
In conclusion, the combined application of single-cell and spatial transcriptomics facilitates the integration of spatial information with single-cell resolution. This approach enables a comprehensive exploration of cellular subpopulation distribution characteristics, developmental differentiation, interactions, and regulatory mechanisms from multiple dimensions.
References:
- Longo S K, Guo M G, Ji A L, et al. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nature Reviews Genetics, 2021, 22(10): 627-644.
- Li Y, Li Z, Wang C, et al. Spatiotemporal transcriptome atlas reveals the regional specification of the developing human brain. Cell, 2023, 186(26): 5892-5909. e22.
- Yang F, Zhao Z, Zhang D, et al. Single-cell multi-omics analysis of lineage development and spatial organization in the human fetal cerebellum. Cell Discovery, 2024, 10(1): 22.

 Sample Submission Guidelines
Sample Submission Guidelines


