
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Single-cell RNA sequencing (scRNA-seq) has revolutionized the field of genomics by providing unprecedented insights into the heterogeneity and complexity of cellular populations. This cutting-edge technology enables the characterization of gene expression profiles at the resolution of individual cells, allowing researchers to delve into the intricate details of cell types, states, and interactions within biological systems. However, the analysis and interpretation of scRNA-seq data pose significant challenges due to the sheer volume and complexity of the data generated.
At CD Genomics, we understand the importance of robust and reliable data analysis in extracting meaningful biological insights from scRNA-seq experiments. Our Single-cell RNA Sequencing Data Analysis Service combines state-of-the-art computational pipelines, expert bioinformatics support, and interactive visualization tools to empower researchers in unraveling the mysteries hidden within the vast landscape of single-cell transcriptomics data. In this article, we will explore the use of scRNA-seq, delve into the comprehensive services we offer, and highlight the advantages of our data analysis service.
Single-cell RNA sequencing has emerged as a transformative technology in the field of genomics, enabling researchers to investigate cellular heterogeneity with unparalleled resolution. Traditional bulk RNA sequencing provides averaged gene expression measurements, masking the underlying diversity within a population of cells. In contrast, scRNA-seq allows the characterization of gene expression profiles at the single-cell level, unraveling the heterogeneity that exists within seemingly homogeneous cell populations.
By analyzing scRNA-seq data, researchers can:
scRNA-seq enables the discovery and classification of previously unknown cell types, providing a comprehensive map of cellular diversity in complex tissues and organs. By identifying unique gene expression signatures, researchers can delineate cell types, define cell subpopulations, and investigate lineage relationships. This knowledge is invaluable in understanding tissue development, disease progression, and cellular responses to external stimuli.
By capturing gene expression profiles of individual cells across different time points or experimental conditions, scRNA-seq allows the reconstruction of cellular trajectories and the inference of dynamic processes, such as cell differentiation, cell cycle progression, and cellular responses to stimuli. Trajectory analysis provides insights into the underlying regulatory networks and molecular events governing cellular transitions, offering a deeper understanding of cellular development and disease mechanisms.
The analysis of scRNA-seq data can uncover interactions between different cell types through the identification of ligand-receptor pairs. By studying the expression patterns of ligands and receptors across cell types, researchers can infer signaling pathways and communication networks within complex biological systems. This knowledge aids in unraveling the interplay between cells and the role of cellular crosstalk in tissue homeostasis, immune response, and disease progression.
At CD Genomics, we offer a comprehensive Single-cell RNA Sequencing Data Analysis Service tailored to meet the diverse needs of researchers in their exploration of cellular heterogeneity. Our service encompasses various stages of data analysis, from quality control and preprocessing to in-depth exploratory analysis and advanced integrative approaches. Let's delve into each aspect of our service:
We understand that the reliability of scRNA-seq data analysis heavily relies on the quality of the input data. Therefore, we employ rigorous quality control measures to assess data integrity, identify potential batch effects, and detect outliers. Through this step, we ensure that downstream analyses are based on high-quality, reliable data. Preprocessing steps include read alignment, gene quantification, normalization, and batch effect correction, which are essential for accurate downstream analysis. CD Genomics utilizes cutting-edge tools and algorithms, such as Seurat, Scanpy, Harmony, and LIGER, to perform these preprocessing steps efficiently and effectively.
During the quality control and preprocessing stage, we generate comprehensive quality control metrics to assess the integrity of the data. These metrics include the number of genes detected per cell, molecular barcodes counts, and the proportion of mitochondrial reads, among others. By visualizing these metrics, researchers can identify potential outliers, low-quality cells, or technical artifacts that may influence downstream analyses. Our data analysis pipeline provides visualizations, such as box plots and scatter plots, to facilitate the assessment of data quality.
Once the data has undergone quality control and preprocessing, we move on to exploratory analysis, which aims to uncover the underlying structure and patterns within the scRNA-seq data. CD Genomics employs dimensionality reduction techniques, such as t-distributed stochastic neighbor embedding (t-SNE), uniform manifold approximation and projection (UMAP), and principal component analysis (PCA), to visualize and cluster cells based on their gene expression profiles.
By visualizing these reduced-dimensional representations, researchers can identify distinct cell clusters corresponding to different cell types or states. CD Genomics provides interactive visualization tools, such as scatter plots and heatmaps, to enable researchers to explore and interact with their data effectively. Additionally, we offer differential gene expression analysis, allowing researchers to identify genes that are uniquely upregulated or downregulated in specific cell clusters.
One of the key goals in scRNA-seq analysis is to accurately identify and annotate cell types within a heterogeneous population. CD Genomics leverages reference-based cell type identification approaches to assign cell types to the clusters identified during exploratory analysis. We utilize established cell type marker genes and publicly available databases, such as CellMarker and Human Cell Landscape, to map the expression profiles of cells to known cell types.
By integrating external knowledge with the scRNA-seq data, we can confidently assign cell type labels to the clusters, providing researchers with a comprehensive understanding of the cellular composition within their samples. This information is vital for unraveling the functional implications of different cell types and their contributions to biological processes or disease phenotypes.
Understanding the dynamic processes and developmental trajectories within a cell population is crucial for comprehending cellular differentiation, maturation, and response to external stimuli. CD Genomics offers trajectory analysis to infer the developmental paths and transitions between different cell states. We utilize algorithms such as Monocle, Slingshot, and PAGA to reconstruct the trajectories and identify crucial branching points or transitional states.
By visualizing the trajectories, researchers can gain insights into the regulatory networks and molecular events that drive cell fate decisions. Our trajectory analysis enables the identification of key genes driving cellular transitions and provides a holistic view of cellular dynamics within the biological system under investigation.
To gain a comprehensive understanding of complex biological systems, it is often necessary to integrate scRNA-seq data from multiple samples or conditions. CD Genomics offers integrative single-cell analysis, which enables the comparison and integration of scRNA-seq data from different experimental conditions or tissues.
Through data integration, researchers can identify shared and distinct cell types, characterize condition-specific gene expression patterns, and unravel the molecular mechanisms underlying phenotypic differences. Our integrative analysis methods, including Seurat, Harmony, and LIGER, allow for seamless integration of scRNA-seq data from diverse sources, promoting a deeper understanding of the biological system under investigation.
Cell-cell communication plays a vital role in the coordination of cellular processes and the regulation of tissue homeostasis. CD Genomics offers ligand-receptor analysis to unravel the intricate intercellular communication networks within scRNA-seq data. Through the identification of ligand-receptor pairs, we can infer potential signaling interactions between different cell types and uncover the underlying molecular mechanisms that drive cellular responses.
CD Genomics utilizes computational approaches, such as CellPhoneDB, scRNASeqNet, and ligand-receptor correlation analysis, to identify potential ligand-receptor interactions within scRNA-seq data. By integrating gene expression information and known ligand-receptor databases, we can pinpoint specific ligand-receptor pairs that are likely to be involved in cell-cell communication.
This analysis provides valuable insights into the potential signaling pathways and communication networks that govern cellular processes within the studied system. By understanding the intercellular communication landscape, researchers can elucidate the role of cell-cell interactions in tissue development, immune responses, and disease progression.
In addition to analyzing gene expression profiles at the single-cell level, CD Genomics also offers spatial transcriptomic analysis, which combines spatial information with gene expression data. Spatial transcriptomics allows researchers to visualize and analyze gene expression patterns within the context of tissue architecture and cellular organization.
By integrating scRNA-seq data with spatial information obtained through techniques such as spatial transcriptomics or in situ sequencing, researchers can gain a deeper understanding of how gene expression varies across different tissue regions or cell types. This analysis provides insights into spatially regulated gene expression, cell-cell interactions within the tissue microenvironment, and the spatial distribution of specific cell types or functional modules.
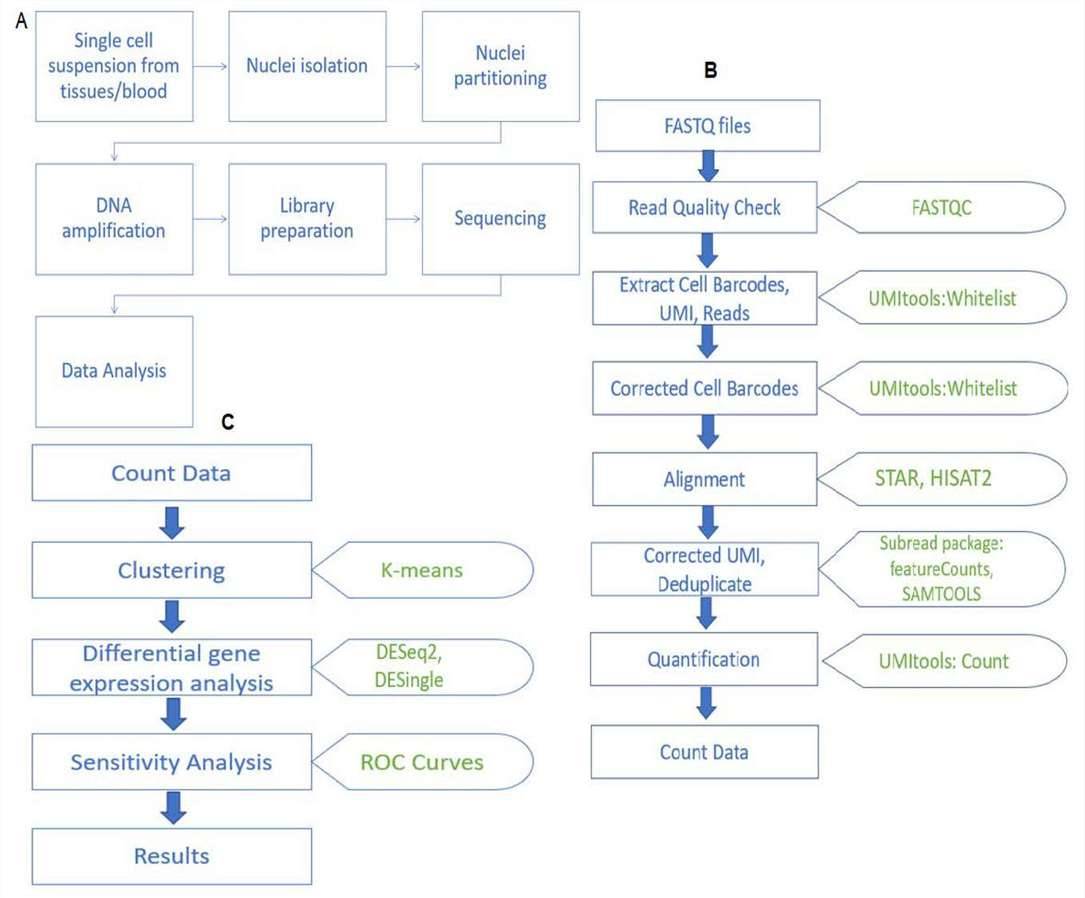
CD Genomics' Single-cell RNA Sequencing Data Analysis Service offers several key advantages that empower researchers in their exploration of cellular heterogeneity:
Expertise and Experience: CD Genomics boasts a team of highly skilled bioinformaticians and data analysts who are well-versed in the complexities of scRNA-seq data analysis. Our experts stay up-to-date with the latest advancements in the field and employ cutting-edge tools and algorithms to deliver reliable and insightful results.
Comprehensive Solutions: Our service covers a wide range of analysis steps, from quality control and preprocessing to advanced exploratory and integrative analyses. By offering a comprehensive suite of analysis tools and methodologies, we ensure that researchers can extract the maximum value from their scRNA-seq data.
Interactive Visualization: CD Genomics provides interactive visualization tools, such as scatter plots, heatmaps, and trajectory plots, which allow researchers to explore their data in a user-friendly and intuitive manner. These visualizations aid in the interpretation of complex biological phenomena and facilitate data-driven decision-making.
Robust Quality Control: We employ stringent quality control measures to ensure the reliability and accuracy of the analyzed data. By identifying potential batch effects, technical artifacts, and low-quality cells, we ensure that downstream analyses are based on high-quality data, leading to more robust and reliable biological insights.
Customizable Analysis: We understand that each research project has unique requirements and objectives. CD Genomics offers customizable analysis pipelines to tailor our services to the specific needs of our clients. Whether it's a focused cell type identification study or a comprehensive integrative analysis, our team works closely with researchers to deliver results that align with their research goals.
Partnering with CD Genomics for scRNA-seq data analysis ensures access to cutting-edge analytical techniques, expert support, and a comprehensive understanding of cellular heterogeneity.
Reference:


