
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Single-cell RNA sequencing(scRNA-seq) is a technique for sequencing the RNA of a single cell and has rapidly developed in recent years. It has advantages in identifying cells at the molecular level and interpreting cellular heterogeneity, and it is now widely applied in medical and biological research.
Early in the 1990s, real-time fluorescence quantitative polymerase chain reaction (PCR) technology for a small number of genes in a single cell appeared [1]. The real scRNA-seq was proposed in 2009 by Tang et al, which sequenced the transcriptome of a small number of mouse primordial germ cells[2]. Since then, a multitude of scRNA-seq technologies have been developed, including STRT-seq[3], CEL-seq[4], SMART-seq[5], and subsequent nanodroplets, picowell technologies and in situ barcoding technologies for high-throughput sequencing. Before 2014, most scRNA-seq platforms were limited to analyzing fewer than 100 cells, and the sequencing cost was high. The emergence of subsequent technologies, such as Drop-Seq[6] and inDrop[7] have enabled high-throughput sequencing. In 2017, the emergence of two commercial high-throughput scRNA-seq platforms, 10X Genomics and BD Rhapsody have made scRNA-seq more widely used, which are still the most widely used platforms today. Since then, scRNA-seq technologies such as Microwell-seq[8] and DNBelab C4[9] have further reduced sequencing costs and increased cell throughput. I divided different technologies into two categories, namely, tag sequencing technology and full-length sequencing technology. Due to the iteration of technologies, sequencing methods proposed at different stages must have their advantages and limitations (Fig.1).
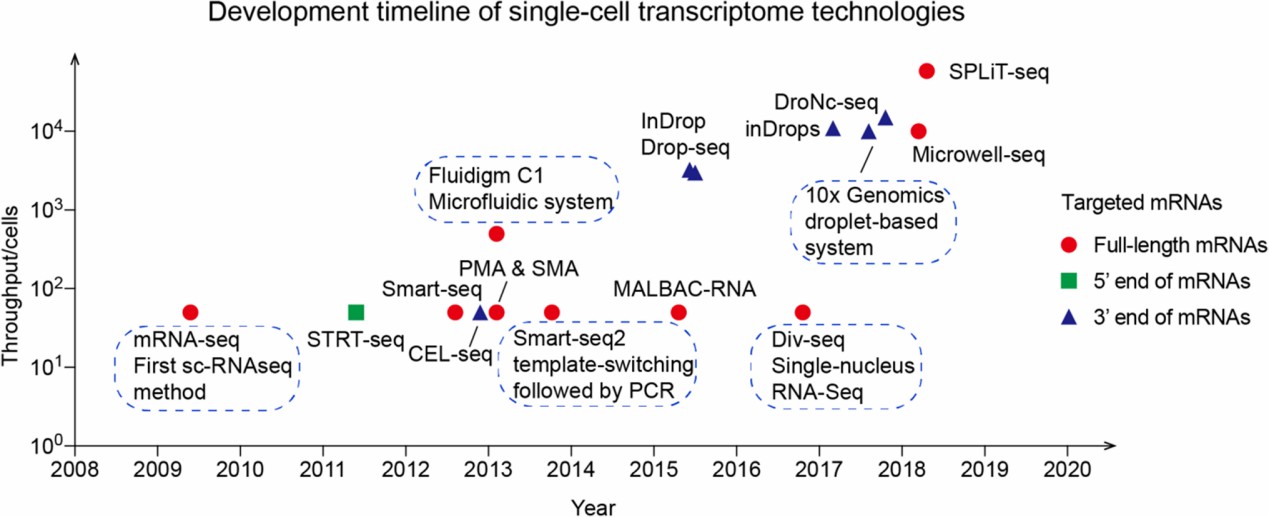 Fig.1 Development timeline of single-cell transcriptome technologies[10]
Fig.1 Development timeline of single-cell transcriptome technologies[10]
Both scRNA-seq and Bulk RNA-seq sequence the transcriptome of a sample and share upstream processes such as RNA extraction, reverse transcription to cDNA, and library preparation for sequencing[11]. Their fundamental difference lies in the sample: scRNA-seq treats each individual cell as a separate sample, whereas Bulk RNA-seq uses a group of cells as one sample. To achieve single-cell isolation and transcriptome library preparation, scRNA-seq requires more advanced instruments, and it generates a significantly larger volume of data, making it more expensive. Due to its high-resolution data, scRNA-seq can detect rare cell types that Bulk RNA-seq might overlook, making it particularly advantageous in studies involving complex cell types, such as the tumor microenvironment (TME) (Fig.2). Each technique has its strengths, and the appropriate method should be chosen based on the specific research question.
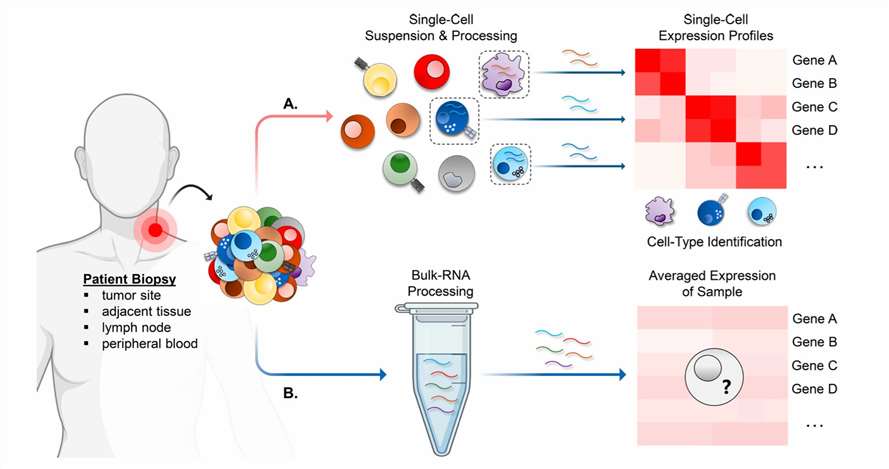 Fig.2 scRNA-seq versus bulk RNA-seq for profiling the TME[12].
Fig.2 scRNA-seq versus bulk RNA-seq for profiling the TME[12].
Accurate and effective analyzing of scRNA-seq data is crucial for scientific research. Therefore, scRNA-seq data analysis methods are continuously being developed and refined. Here, we will outline the typical steps in single-cell data analysis (Fig.3).
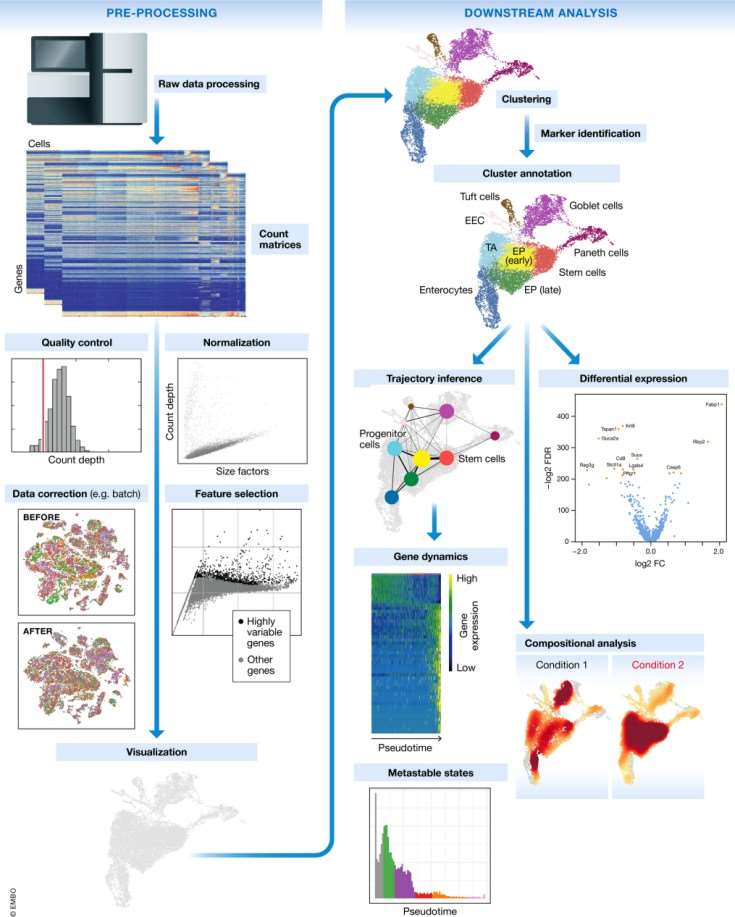 Fig.3 Schematic of a typical single-cell RNA-seq analysis workflow[13].
Fig.3 Schematic of a typical single-cell RNA-seq analysis workflow[13].
After obtaining scRNA-seq data, the first task is to perform quality control. This step includes assessing the quality of the sequencing reads, filtering out low-quality cells, and removing ambient RNA contamination. Quality control tools designed for bulk RNA-seq data, such as Trimmomatic[14], Fastp[15], and Cutadapt[16], are also suitable for preprocessing scRNA-seq raw data. Typically, when processing scRNA-seq data downloaded from the web, I use TrimGalore, a tool that integrates Cutadapt and FastQC (https://github.com/FelixKrueger/TrimGalore). FastQC is another tool published on GitHub that can assess the quality of sequencing reads (https://github.com/s-andrews/FastQC).In the specific analysis process, I will use FastQC to first assess whether the raw sequencing data requires further quality control. If the assessment shows anomalies such as the presence of adapters, low-quality reads, or an excessive number of duplicated fragments, I then use TrimGalore for data cleaning. After quality control of the reads, the next step involves aligning the sequencing reads to the reference genome and generating a quantitative data matrix. For this process, I use integrated alignment and quantification tools such as CellRanger, developed by 10X Genomics[17]. It provides a comprehensive solution for generating single-cell transcriptome data matrices from raw sequencing data, offering a reliable data processing method for single-cell research.
After obtaining the data matrix, we typically need to identify doublets and empty cells. Abnormally high numbers of reads and genes may indicate the presence of doublets. I often use doublet detection tools such as DoubletDecon[18] and DoubletFinder[19] to exclude doublets. Additionally, a high proportion of mitochondrial genes and a low number of genes usually suggest poor cell quality. Ambient RNA contamination refers to RNA present in the single-cell suspension that is detected along with the cell's internal RNA during droplet formation, even though it is not actually present in the cell. To remove this contamination, I use DecontX[20] to quickly predict and correct for ambient RNA contamination, and then proceed with downstream analysis using the corrected data matrix.
Normalization is the first step in scRNA-seq matrix data processing and directly impacts the accuracy of downstream analysis results. A commonly used normalization method assumes that each cell has the same initial number of transcripts and simply normalizes the data to counts per million (CPM). Log normalization of molecular barcodes based data, as implemented in Seurat, is one of the most widely used methods [21]. Other methods, such as sctransform[22], BayNorm[23], and SCnorm[24], can also be used to normalize scRNA-seq data. For full-length sequencing methods like SMART-seq, transcript length is usually taken into account during data processing.
Currently, scRNA-seq data has become highly abundant, and effectively integrating data from different batches has become a new challenge. Batch effects imply technical differences that arise when samples come from different batches, which may result from factors such as different time points, different operators, varying scRNA-seq protocols, or inconsistencies in the sequencing samples. Therefore, several methods have been developed specifically to eliminate batch effects in scRNA-seq data. Commonly used data integration methods include Seurat[21], MNN[25], Harmony[26], and Conos[27]. I most frequently use the CCA algorithm included in Seurat and Harmony. Harmony eliminates batch effects while preserving the biological differences between the two samples, whereas CCA applies a stronger correction, which may potentially erase biological differences between the samples.
In single-cell data analysis, the accurate identification and annotation of cell types is a critical step for all downstream analyses. This process includes steps such as feature selection, dimensionality reduction, clustering, and annotation. The first step in dimensionality reduction for scRNA-seq data is feature selection, where the dataset is filtered to retain only the genes that contribute significantly to the data's variability. These retained genes are known as highly variable genes[28] (HVGs).The number of HVGs typically ranges from 1,000 to 5,000 and needs to be adjusted based on the complexity of the dataset. After selecting the HVGs, the dimensionality of the scRNA-seq expression matrix needs to be further reduced, describing the data with far fewer dimensions than the number of genes, usually in two or three dimensions. Common dimensionality reduction methods now include both linear and nonlinear approaches, with Principal Component Analysis[29] (PCA) being the most popular linear method. PCA analysis is typically used as a preprocessing step for nonlinear dimensionality reduction, and the number of principal components (PCs) selected for the next step can affect the interpretation of subsequent results. Nonlinear dimensionality reduction methods allow for the visualization of data in two or three dimensions. The most commonly used methods include t-distributed stochastic neighbor embedding[30] (t-SNE) and uniform manifold approximation and projection[31] (UMAP).The purpose of cell clustering is to group cells based on the similarity of their gene expression patterns, in order to obtain biologically meaningful subpopulations. Clustering based directly on matrix distances is an unsupervised machine learning process, with k-means clustering being a widely used method[25]. Accurate annotation of the clusters obtained from clustering is a key step in scRNA-seq data analysis. Currently, this process is typically achieved through both manual annotation and automatic annotation methods. Manual annotation involves matching the characteristic genes of each cluster with published literature and databases, then assigning biologically meaningful cell identities to the clusters. For example, the table below summarizes some hallmark genes for various cell types (Table 1).
| Annotation | Gene signature | Refs |
| Naive T cells | CD45RA, IL7R, CD27, CCR7, CXCR5 | [32] |
| Cytotoxic T | GZMA ,NKG7 ,GZMB ,GZMK ,FASLG,ITGA1 ,CXCR6 | [33] |
| Proliferation T | RRM2 ,TK1 ,CENPF ,CENPM ,MKI67 ,MCM4 | [34] |
| Early-stage T | CCR7, IL7R , TCF7 ,CD28 | [35] |
| Central memory cells | CCR7, SELL, GPR183, GZMK | [36] |
| CCR7 ,IL7R , SELL ,TCF7 | [37] | |
| Exhausted T | CTLA4 ,LAG3 ,HAVCR2 ,TRPS1, PDCD1, FABP5, TRPS1, CREM, CEBPD | [35] |
Table.1 Hallmark Genes for Common T Cell Types
Multi-faceted interpretation of annotated cells is part of the downstream analysis of scRNA-seq data. Common analyses include changes in cell composition, differential analysis at the gene level, trajectory inference, and analysis of cell-to-cell communication.
Changes in cell composition refer to the variation in the proportions of various cell types between different groups (e.g., control and experimental groups). For example, there may be an increase in the proportion of neutrophil progenitor cells in the bone marrow of tumor-bearing wild-type mice[38]. Typically, visualizations such as bar charts, area plots, or pie charts are used to present the changes in cell proportions between different groups.
Differential analysis at the gene level encompasses several aspects, including the calculation of differentially expressed genes between groups, gene set enrichment analysis, and inference of transcription factor regulatory networks. Through these analyses, gene expression differences under varying contexts can be characterized, revealing changes at the gene level. To better interpret the biological significance of these genes, researchers them based on common biological processes. These gene sets typically come from databases such as MSigDB[39] and Gene Ontology[40], as well as pathway databases like KEGG[41] and Reactome[42]. Gene function is not carried out independently; transcription factors play a crucial role in the regulation of gene expression. By using transcription factor regulatory network analysis, interactions between transcription factors and other genes can be revealed. Currently, there are specialized tools for this analysis based on scRNA-seq, such as SCENIC[43].
To characterize the continuous changes between cells, dynamic models of gene expression need to be constructed, and these methods are referred to as trajectory inference (TI). TI arranges cells based on transcriptional changes, and this pathway is considered as pseudotime in cell development[44]. Since the creation of Monocle[45] for TI, various algorithms have been rapidly developed.
Cell-to-cell communication refers to interactions mediated by receptor-ligand or other auxiliary factors, which are crucial for biological processes such as cell development and disease progression. Predicting cell communication requires the scRNA-seq expression matrix and known receptor-ligand pairing relationships. Currently, commonly used tools for this purpose include CellphoneDB[46], CellChat[47], and NicheNet[48].
You may interested in
Learn More
The applications of scRNA-seq are extensive, covering multiple fields of biology and medicine. Here, I summarize its applications in oncology, immunology, developmental biology, and neuroscience.
scRNA-seq has been widely used in human tumor research, including studies on tumor heterogeneity, TME, and cell interactions. Tumor heterogeneity encompasses differences between tumors as well as variations within a single tumor. Accurately identifying tumor heterogeneity plays a crucial role in the diagnosis and treatment of diseases[49]. Durante et al. discovered previously unrecognized sub-clonal genomic complexity and transcriptional states in melanoma[50]. TME is a complex ecosystem composed of cancer cells, various non-malignant cells, extracellular matrix, secreted factors, and tumor vasculature. Zheng et al. revealed that exhausted T and NK cells, Treg cells, selectively activated macrophages, and tolerogenic dendritic cells dominate the TME in esophageal cancer[51]. The crosstalk between macrophages and Tregs contributes to potential immune suppression within the TME. Cell-to-cell interactions primarily focus on the interactions between malignant cells and the TME or cells originating from the TME. Wei Zhuo et al. discovered a mechanism in which Cadherin 11-mediated signaling between gastric cancer cells and fibroblasts promotes gastric cancer metastasis[52].
scRNA-seq can be used to comprehensively analyze different cell types in the immune system, revealing functional differences among various immune cells in health and disease states. Among all immune cells, scRNA-seq studies on T cells are the most numerous, as disease is often associated with changes in T cell status. By conventional methods, classical T cell subsets have been identified, including naive, memory, and effector T cells. However, scRNA-seq of these classical subsets has revealed new findings of exhausted[53], cytotoxic, and immunosuppressive T cells[54]. In addition to identifying new cell types, scRNA-seq can also analyze the impact of diseases on immune cells and help understand disease mechanisms. For example, myeloid-derived suppressor cells, which differentiate from Granulocyte-Monocyte Progenitors, accumulate in large numbers within the TME[55]. These cells infiltrate tumors and directly promote angiogenesis and metastasis, while also suppressing immune responses and reducing the effectiveness of immunotherapy.
scRNA-seq can capture the gene expression profiles of cells at different stages of development, helping to construct developmental trajectories from stem cells to specialized cells and revealing gene regulatory networks during the developmental process. Lars M. Steinmetz et al. conducted a single-cell analysis of human bone marrow hematopoietic stem and progenitor cells (HSPCs), showing that the acquisition of lineage-specific fates is a continuous process[56]. During embryonic development, scRNA-seq can be used to trace cell fates and study how different tissues and organs gradually form during early development. The team at the Sanger Institute utilized scRNA-seq of thymic tissue during the embryonic stage to reveal the development of the human thymus and the maturation process of T cells. Their research found that the corresponding changes in thymic stromal cells reflect trends in T cell development[57].
The brain contains various types of neurons, each differing in morphology, function, and gene expression. scRNA-seq can identify and classify these neurons, revealing their distinct gene expression patterns[58]. In addition to neurons, the brain also contains a large number of glial cells (such as astrocytes, oligodendrocytes, and microglia). scRNA-seq can reveal the gene expression characteristics of these non-neuronal cells, helping to understand their roles in neural support, protection, and repair. Elizabeth et al. conducted an analysis of human cortical samples based on single-cell RNA sequencing and purified viable microglial cell subpopulations from these samples, discovering that some cell subpopulations were enriched with genes and RNA markers associated with neurodegenerative diseases[59].
In 2013, scRNA-seq was named Technology of the Year by Nature Methods[60], and in 2018, Science named scRNA-seq Breakthrough technology of the Year[61], whose rapid development has greatly broadened our understanding of cell heterogeneity and cell function. The rapid development of this technology has greatly expanded our understanding of cell heterogeneity and function. The instruments for single-cell sequencing are continuously being iterated and upgraded, while the development of data processing tools is also progressing rapidly, facilitating the widespread application of this technology in biology and medicine. scRNA-seq is evolving towards lower costs, higher throughput, and multi-omics capabilities, and it is expected to see even broader use in the fields of biology and pharmaceuticals in the future.
References:


