As molecular biology continues to advance, genetic technologies undergo constant updates and improvements. In 1977, Sanger's invention of the dideoxy chain termination method marked a milestone in the field of sequencing, heralding a new era in nucleic acid analysis and ushering in the automation age for genetic sequencing. Subsequently, the emergence of second-generation sequencing technologies, such as Next-Generation Sequencing (NGS), further enhanced the efficiency and capabilities of sequencing techniques.
Confronted with vast amounts of genetic information akin to the twinkling stars in the night sky, the complexity of genetic inheritance poses significant challenges. While harnessing the power of NGS technology, ensuring the quality of NGS sequencing has become an imperative issue that demands our immediate attention.
In this article, we delve into the significance of NGS validation, the workflow of NGS confirmation, and the reasons why validation is imperative in genomic analysis.
What is NGS Validation?
NGS validation is the systematic evaluation of the accuracy, precision, and reliability of NGS assays and platforms. This process entails thorough testing to guarantee the consistency and reproducibility of results produced by NGS technologies. Validation encompasses a range of parameters, including analytical sensitivity, specificity, precision, and accuracy.
Assessment of Analytical Sensitivity and Specificity
Analytical sensitivity denotes the capacity of an NGS assay to identify genuine positive variants, particularly those existing at low allelic frequencies. Conversely, analytical specificity relates to the assay's capability to precisely discern true negative outcomes while mitigating false positives.
Precision and Accuracy
Precision quantifies the extent of reproducibility or repeatability of outcomes within a singular assay or across multiple assays. Conversely, accuracy delineates the degree to which the produced NGS data align with the authentic genomic sequence.
NGS Confirmation Workflow
The workflow for confirmation in NGS proves vital in preserving the dependability and precision of the genomic data amassed from NGS platforms. This intricate process encompasses several phases that are directed towards verifying the detected variants using complementary methodologies, such as Sanger sequencing. The succeeding discourse further examines the individual stages of the NGS confirmation workflow:
Variant Identification by NGS
Commencing with the identification of variants by NGS, these platforms deploy advanced, high-throughput sequencing technologies which yield copious amounts of sequencing data distilled from genomic DNA samples. This incipient phase in the confirmation workflow encompasses variant identification, herein the deployment of bioinformatics instruments is essential for scrutinizing the profuse raw sequencing data and discerning genetic variations, inclusive of SNVs, insertions, deletions, and structural deviations.
This process encases the alignment of sequencing interpretations onto a reference genome, distinguishing variants, and commenting on their functional and clinical connotations.
Selection of Variants for Confirmation
Not all variants detected through NGS necessitate validation via orthogonal approaches. Variants surpassing predefined quality benchmarks, including depth of coverage, variant allele frequency, and variant calling precision, may be regarded as robust and exempt from confirmatory procedures. Nevertheless, variants failing to meet these quality standards or possessing clinical relevance may necessitate additional validation via confirmatory assays, such as Sanger sequencing.
Sanger Sequencing Confirmation
Sanger sequencing, commonly referred to as chain termination sequencing, stands as a classical DNA sequencing technique esteemed as the benchmark for corroborating genetic variants pinpointed through NGS. During the validation phase, targeted segments housing the identified variants undergo amplification via PCR utilizing primers tailored for this purpose. Subsequently, the amplified fragments undergo Sanger sequencing, wherein the incorporation of chain-terminating dideoxynucleotides facilitates the sequencing of individual DNA fragments. The resultant sequencing data undergo meticulous analysis to decipher the nucleotide sequence, thereby affirming the existence of the pertinent variant.
Data Analysis and Interpretation
Upon completion of Sanger sequencing, the resultant data undergo comparison with the original NGS dataset to evaluate concordance. Any inconsistencies between these datasets undergo thorough scrutiny to ascertain the precision of the NGS variant identifications. Confirmation of the variant by Sanger sequencing enhances confidence in the fidelity of the NGS outcomes. Conversely, discrepant findings prompt further inquiry to elucidate potential factors such as sequencing errors, alignment anomalies, or sample contamination.
By adhering meticulously to the NGS confirmation protocol, laboratories uphold the integrity and precision of genomic data emanating from NGS platforms. This meticulous validation regimen plays a pivotal role in underpinning clinical decision-making, delineating personalized therapeutic approaches, and upholding the pinnacle of patient care standards.
Why is NGS Validation Necessary?
Despite the widespread adoption and continual refinement of standard procedures for NGS, performance validation remains an indispensable step. This is primarily because performance validation ensures the accuracy, reliability, and consistency of NGS in practical applications, thereby safeguarding the quality and safety of clinical diagnostics and treatment.
1. Multiple Factors Affecting Detection Results
The implementation of NGS technology is not only constrained by factors such as personnel proficiency, laboratory conditions, instrument and reagent quality, and standardization of operational procedures, but also susceptible to introducing errors at each step due to its encompassment of numerous stages, thereby significantly influencing the final detection outcomes. Specifically, potential issues may include:
- During the library construction process, improper handling may lead to failure in the connection of tags or adapters.
- Discrepancies or errors in annotation stages may result in analytical bias.
- Automation extraction techniques may occasionally induce cross-contamination between samples, affecting result accuracy.
- Inefficient probe capture may impede the effective capture of target sequences.
- Identification errors during the tagging process may result in misinterpretation of data.
- Improper parameter settings or excessive data filtering during the data analysis phase may impact result reliability.
- Errors in the variant detection process may lead to misjudgments of variant types.
- Optical biases in the sequencing instrument's optics may provoke sequencing base quality issues, further influencing sequencing accuracy.
Given the aforementioned reasons, different processes and laboratories often exhibit certain disparities in sequencing quality and information analysis outcomes when implementing NGS technology. Hence, in practical applications, stringent control of operational quality at each stage is imperative to ensure the accuracy and reliability of data.
2. Additional Methods to Ensure NGS Analysis and Clinical Utility
The capability of NGS testing to identify variants of significant clinical relevance, as well as those whose clinical significance remains unclear, necessitates notable variations in the clinical validation requirements for each variant. Moreover, the primary anticipated utility of NGS testing is focused on specific patient cohorts, drugs, or disease domains. However, as evidenced by research findings from the National Cancer Institute's Molecular Analysis for Therapy Choice (MATCH) program, the selection of therapeutics or agents for treatment should be based on specific molecular findings revealed by targeted NGS analysis, rather than solely relying on cancer type classification.
The rapid advancement of targeted enrichment techniques and NGS has profoundly transformed clinical diagnostic testing. It enables the simultaneous analysis of all genes involved in disease phenotypes at a lower cost, surpassing conventional capillary electrophoresis sequencing, thereby becoming the preferred method for most high-throughput diagnostic laboratories. Targeted NGS panels have become the frontline detection method for various genetic disorders. With the widespread adoption of NGS, numerous diagnostic laboratories have introduced panels for hereditary cancer susceptibility genes, capable of detecting various characteristic genes associated with hereditary cancer.
There are numerous panels available for testing known gene mutations associated with hereditary cancer. The accuracy of these tests may be influenced by various factors, including targeted enrichment platforms, sequencing technologies, employed bioinformatics workflows, and expertise in variant interpretation.
3. The Significance of NGS Oversight
True to their commitment towards ensuring the accuracy and reliability of genomic data sourced from NGS technologies, esteemed entities such as the Centers for Disease Control and Prevention (CDC), the American College of Medical Genetics and Genomics (ACMG), and the American Society for Clinical Pathology (ASCP) have collectively established a set of meticulous guidelines. These normative instructions pertain to the effective validation of NGS methodologies, diligent oversight of analytical processes, as well as comprehensive reporting of identified variants. Relative to conventional molecular detection methods, clinical testing via NGS characteristically poses significantly complex aspects, hence underscoring the critical necessity of robust validation processes for NGS-derived results.
4. Challenges in Providing Precision Medicine Guidance
In an epoch progressively marked by precision medicine and personalised therapies, genetic testing assumes an essential role in steering subsequent therapeutic strategies tailored to an individual patient's needs. The timeliness and accuracy of such diagnostic findings offer clinicians cogent evidence to inform the selection of patients best suited to specific treatment strategies, thus heightening treatment efficacy. Despite the appreciable advantages inherent to NGS - notably its capacity to identify a vast range of variations within the human genome - it yet remains deficient in accomplishing individual validation and scrutiny for every posited variation. Given the heterogeneities in selection criteria across various pathological entities, this shortcoming renders NGS technology somewhat inadequate in delivering precise therapeutic guidance during critical and non-replicable clinical scenarios requiring urgency.
You may interested in
Learn More
Next-generation Sequencing
Next-generation sequencing, also known as high-throughput sequencing, has evolved from DNA sequencing technologies based on PCR and gene chips.
Second-generation sequencing introduced reversible termination at the ends, thereby enabling sequencing by synthesis. In second-generation sequencing, the DNA sequence is determined by capturing the special labels (usually fluorescent molecular tags) carried by the newly added bases during the DNA replication process. Existing technology platforms mainly include Roche's 454 FLX and Illumina's Miseq/Hiseq.
In second-generation sequencing, individual DNA molecules must be amplified into gene clusters composed of identical DNA and then undergo synchronous replication to enhance the fluorescence signal intensity for reading the DNA sequence. As the read length increases, the synergy of gene cluster replication decreases, leading to a decline in base sequencing quality. This strictly limits the read length of second-generation sequencing (not exceeding 500 bp), thus giving it the characteristics of high throughput and short read length.
Sanger Sequencing
Sanger's dideoxy chain termination sequencing method is the mainstream of first-generation sequencing, often referred to as Sanger sequencing. In four DNA synthesis reaction systems, a certain proportion of ddNTP labeled with effective radioactive isotopes is added (the labeled ddNTP technology and corresponding detection technology are also continuously improving and iterating). Through gel electrophoresis and autoradiography, the DNA sequence of the test molecule can be determined based on the position of the electrophoretic band.
Sanger sequencing is simple to operate, mature in method, with a longer read length, and extremely high accuracy. It is the gold standard for sequencing and the international gold standard for genetic sequence analysis. The large-scale sequencing technology used in the Human Genome Project (HGP), proposed in 1985 and formally launched in 1990, is based on the Sanger method.
Sanger sequencing for NGS validation can effectively monitor the quality of NGS data and achieve technical complementation and improvement.
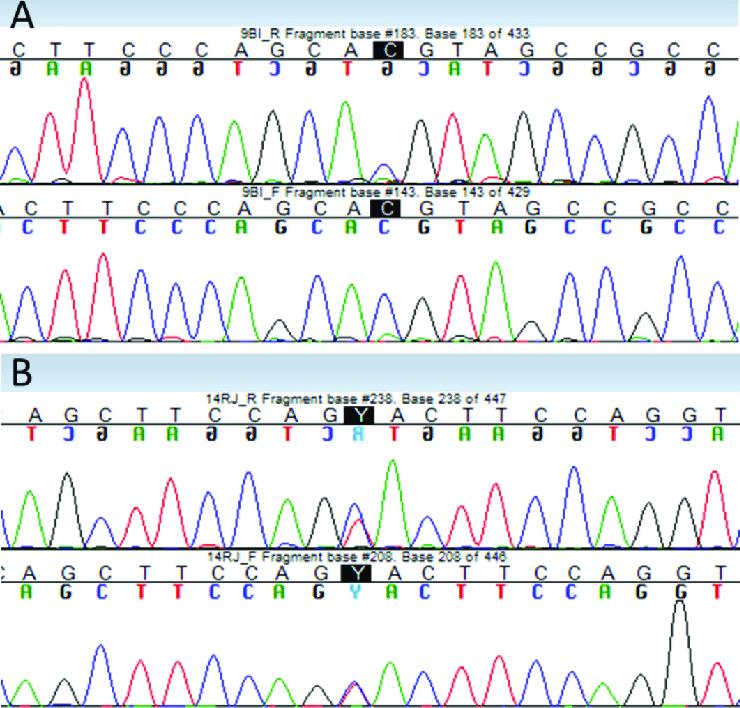 Results of validation of next-generation sequencing (NGS) variants by Sanger sequencing. Verified bases are highlighted in black (A) homozygote variant PRNP gene Val129Met. (B) Heterozygous variant in FGA gene Thr331Ala. (Zuzana Chyra Kufova et al,. 2018)
Results of validation of next-generation sequencing (NGS) variants by Sanger sequencing. Verified bases are highlighted in black (A) homozygote variant PRNP gene Val129Met. (B) Heterozygous variant in FGA gene Thr331Ala. (Zuzana Chyra Kufova et al,. 2018)
Case Study of Next-Generation Sequencing Confirmation
The following article delves primarily into the issues of accuracy and reliability concerning the detection results of hereditary cancer through NGS technology. It examines the circumstances under which variant results obtained from NGS sequencing necessitate further confirmation through Sanger sequencing.
Title: Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing
Q: What types of variants underwent Sanger sequencing in the study?
In this study, Sanger sequencing was conducted for all non-polymorphic variants, totaling 7845. Among these, 1.3% were identified as NGS false positives, primarily located in complex genomic regions such as AT-rich regions, GC-rich regions, single nucleotide repeat sequences, and pseudogene regions. Validation of experimental results was performed by simulating a zero false positive quality score threshold, which revealed a reduction in false positive rates but also a decrease in sensitivity. Therefore, to maintain the highest sensitivity and specificity, Sanger sequencing confirmation of variants identified by NGS is necessary.
Q: When is Sanger sequencing validation necessary during NGS Panel testing?
Sanger sequencing validation is warranted when non-polymorphic variants detected by NGS fail to meet conservative quality thresholds, such as minimum coverage and heterozygosity ratio, for reporting. Additionally, in complex regions such as AT-rich regions, GC-rich regions, single nucleotide repeat sequences, and pseudogene regions, Sanger confirmation is recommended to enhance sensitivity and specificity.
Q: How to set appropriate quality thresholds for NGS Panel testing?
Firstly, regarding coverage, ensuring a minimum coverage of 100x is essential to guarantee the accuracy and reliability of detection results. Such coverage adequately meets the demands of detection and mitigates the risk of errors resulting from insufficient coverage.
Secondly, concerning heterozygosity ratio, for variants with higher heterozygosity, it is advisable to moderately lower the threshold for heterozygosity ratio. For instance, setting the heterozygosity ratio to 40% or higher helps capture more variant information, thereby enhancing detection sensitivity.
Furthermore, in the selection of genomic regions, it is recommended to employ higher quality thresholds for certain complex genomic regions such as AT-rich regions, GC-rich regions, single nucleotide repeat sequences, and pseudogene regions. This aids in improving detection sensitivity and specificity while reducing interference and misjudgments arising from region-specific characteristics.
It is important to note that the aforementioned threshold settings are based on empirical conclusions derived from extensive sample data and are not immutable standards. In practical applications, flexibility in adjusting and optimizing these thresholds is necessary based on specific sample characteristics, experimental conditions, and detection requirements to ensure the accuracy and reliability of detection results.
Sanger Sequencing Results Contradict NGS Sequencing Results
The basic process of Sanger sequencing includes: first, accumulating a sufficient quantity of DNA fragments from the target region through conventional PCR; next, using the resulting DNA fragments as templates for amplification with ddNTPs to produce a series of single-stranded DNA fragments of varying lengths; finally, sequencing is performed. It is evident that the accuracy of PCR amplification directly influences the final sequencing results. Any deviation during the initial PCR amplification step can lead to inaccurate sequencing results. The main factors affecting the accuracy of PCR results are the specificity and accuracy of the amplification primers, highlighting the crucial importance of primer design.
Generally, primer design should adhere to the following principles:
(1) Primer length is typically between 18-27bp; excessively long primers can lead to extension temperatures exceeding 74°C, unsuitable for Taq DNA polymerase reactions.
(2) The GC content of primers is typically between 40%-60%, with an optimal range of 45-55%. Both excessively high or low GC content is unfavorable for triggering reactions. The GC content and Tm values of upstream and downstream primers should be close, with the Tm value generally not exceeding 5°C.
(3) The 3' end of the primer generally does not contain an A base, and should not contain more than three consecutive bases, such as GGG or CCC, which may cause PCR reaction failure.
(4) The bases of the primer are preferably randomly distributed, and there should not be four consecutive complementary bases between the primer itself and between primers.
(5) Use UCSC In-Silico PCR retrieval to confirm primer specificity.
(6) The distance between the primer's chromosomal position and the detection site should be between 80bp and 800bp.
Exploring Discrepancies between Sanger and NGS Sequencing Results
In addressing instances where Sanger sequencing results diverge from those obtained via NGS sequencing, following a preliminary validation of the reliability of NGS sequencing data, our primary recommendation is to conduct a thorough verification of the specificity of the primers utilized in Sanger sequencing. Through meticulous scrutiny of NGS data, it is imperative to examine whether the binding sites of Sanger amplification primers harbor SNVs, as these variants may interfere with the amplification process and result in disparities between the outcomes. Should such variant sites be identified, a redesign of primers specific to the sample in question becomes necessary to rectify the inconsistency between Sanger and NGS sequencing results.
Indeed, aside from rare instances where SNV mutations lead to aberrant Sanger sequencing outcomes, the human body harbors a vast array of SNP sites. These SNP sites, abundant within the genome, represent the most prevalent form of inheritable variation in humans, encompassing over 90% of known polymorphisms. In typical scenarios, there may exist one SNP site per five hundred to one thousand base pairs, with their total count surpassing three million. Hence, the presence of these SNP sites within primer sequences may similarly induce amplification biases, thereby compromising the accuracy of sequencing results. Taken together, the issue of primer binding biases in Sanger sequencing may be widespread and warrants considerable attention.
Consequently, when designing Sanger primers, a conscious effort must be made to proactively avoid potential SNV and SNP sites. To circumvent SNP sites, querying the UCSC website for the presence of such sites within primer sequences is advisable. In instances of discordance between Sanger and NGS sequencing results, particular attention should be given to whether primers align with SNV regions.
Summary
It is noteworthy that even the acclaimed "gold standard" Sanger detection technique may, in extremely rare cases, yield false positive and false negative results. Although such occurrences are infrequent, they demand our utmost vigilance. Hence, in practice, no single detection technique is flawless. To ensure the accuracy and reliability of variant detection, it is advisable to employ a complementary array of detection methodologies to collectively address issues and pursue the most authentic representation of variant occurrences.
References:
- Mu W, Lu HM, Chen J, et al. Sanger Confirmation Is Required to Achieve Optimal Sensitivity and Specificity in Next-Generation Sequencing Panel Testing. J Mol Diagn. 2016 Nov;18(6):923-932. doi: 10.1016/j.jmoldx.2016.07.006. Epub 2016 Oct 6. PMID: 27720647.
- Rehm HL, Bale SJ, Bayrak-Toydemir P, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med 2013, 15:733-747.
- C W Dieffenbach 1, T M Lowe, G S Dveksler. General concepts for PCR primer design.1993 Dec;3(3):S30-7. doi: 10.1101/gr.3.3.s30.

 Sample Submission Guidelines
Sample Submission Guidelines


