
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Messenger RNA sequencing (mRNA-Seq) is an important high-throughput technique used to analyze gene expression in detail. This approach is extensively utilized in examining gene expression patterns, transcript variants, and RNA modifications. By sequencing cellular mRNA, mRNA-Seq provides deep insights into gene activity changes under various physiological and pathological conditions. Such revelations are invaluable for researchers aiming to decipher the complex mechanisms of gene expression regulation. mRNA-Seq plays an important role in several scientific fields, including epigenetics, oncology, pharmacology, and personalized medicine.
The core principle of mRNA-Seq lies in leveraging high-throughput sequencing technologies to perform an exhaustive quantitative evaluation of all genes within a transcriptome. This method affords detailed insights into gene expression levels, alternative splicing events, transcriptomic structural changes, and the functional significance of non-coding RNAs. When contrasted with traditional microarray methods, mRNA-Seq exhibits superior sensitivity and generates a richer tapestry of data, adept at unveiling novel transcripts and alternative splicing forms. As such, mRNA-Seq stands as an indispensable tool for the intricate exploration of gene function and regulatory networks.
Experimental Workflow:
The procedure for mRNA-Seq encompasses several essential stages:
1. RNA Extraction and Purification: The process begins with the extraction of total RNA from cellular or tissue samples. As ribosomal RNA (rRNA) makes up over 90% of the total RNA content, strategies such as Oligo(dT) bead-based enrichment are routinely applied to specifically isolate mRNA, which is characterized by polyA tails. Enhancing mRNA specificity at this juncture is crucial to improving the experiment's efficiency and precision.
2. RNA Fragmentation and Reverse Transcription: Isolated mRNA is subsequently fragmented into smaller pieces, usually using chemical methods like heating or enzymatic digestion. Following fragmentation, reverse transcription is conducted with reverse transcriptase to convert mRNA fragments into complementary DNA (cDNA).
3. cDNA Library Construction: The resulting cDNA is modified by appending specific adaptor sequences necessary for sequencing. Depending on the research focus, strand-specific library construction might be employed, which is particularly important for analyzing specific transcript variants.
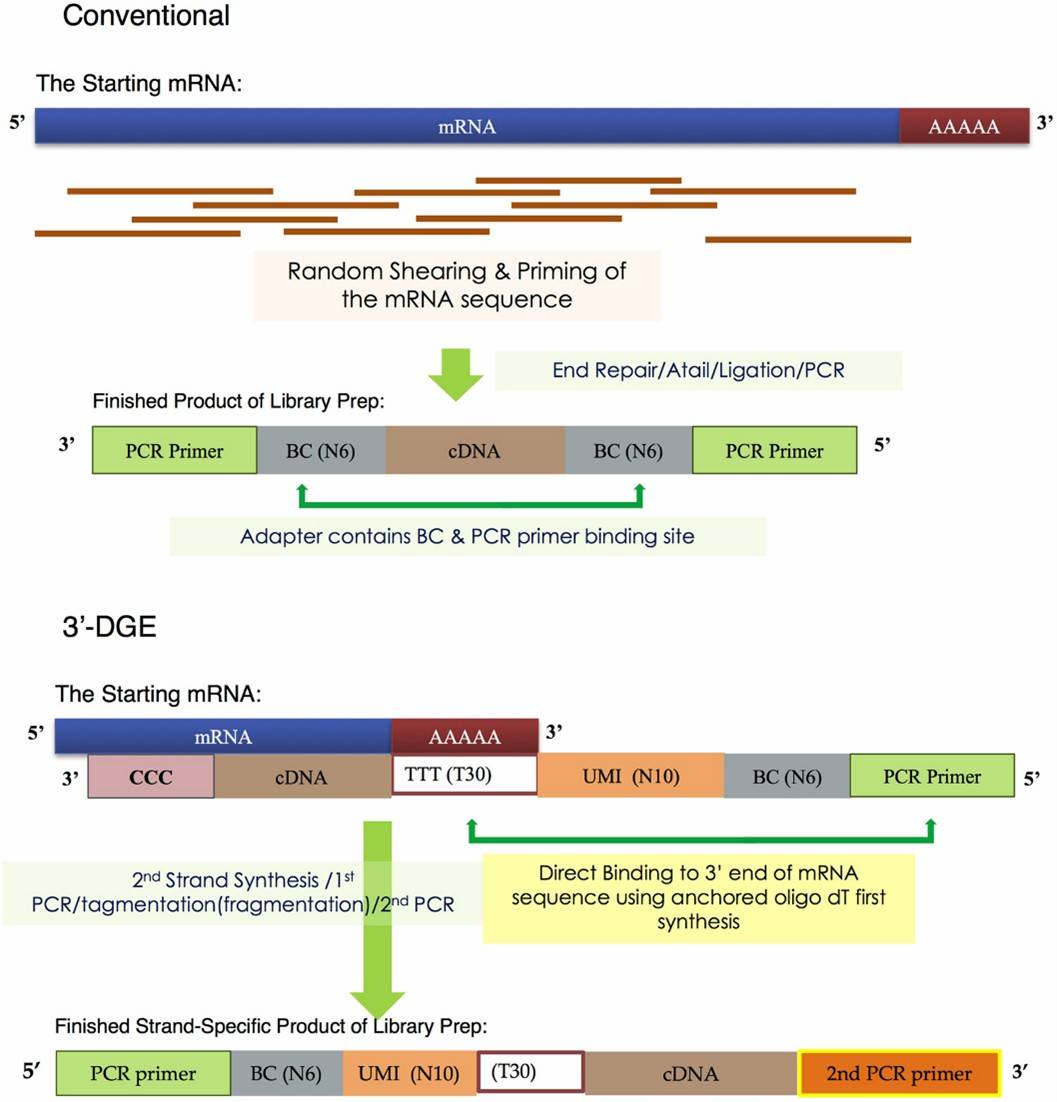 Schematic of Library Construction Differences between Conventional and 3'-DGE mRNA Sequencing. (Xiong, Y., et al., 2017)
Schematic of Library Construction Differences between Conventional and 3'-DGE mRNA Sequencing. (Xiong, Y., et al., 2017)
4. High-throughput Sequencing: The cDNA library is then subjected to high-throughput sequencing using platforms such as Illumina, PacBio, or Oxford Nanopore, to capture the sequence information of the mRNA fragments.
5. Data Analysis: Comprehensive analysis of the sequencing data follows, involving alignment, annotation, and interpretation to glean insights into gene expression levels, splice variations, and transcript diversity. This analysis is accompanied by essential processes such as quality control, data normalization, and differential expression analysis.
Technical Overview
mRNA-Seq is a powerful high-throughput technology with significant advantages but also some challenges. Maintaining RNA purity and integrity is crucial to avoid degradation or contamination that could affect the results. Utilizing high-quality RNA samples is imperative for obtaining precise sequencing data. The reliability of data depends on careful consideration of factors such as replicates, control group configurations, and sample selection. Furthermore, choosing the appropriate sequencing depth is critical; inadequate depth might miss low-abundance transcripts, while excessive depth can lead to unnecessary costs. Data normalization is vital for comparisons across different samples, helping to correct biases stemming from variations in sequencing depth.
In conclusion, mRNA-Seq provides valuable insights into gene expression. However, achieving reliable results requires careful planning and optimization of both experimental methods and data analysis.
Services you may interested in
Want to know more about the details of mRNA sequencing? Check out these articles:
mRNA is a single-stranded RNA molecule transcribed from DNA, carrying genetic information from DNA to ribosomes, where protein synthesis occurs in the cytoplasm. This sequence encodes the instructions for protein production, and its structure is crucial for the stability and translation efficiency of gene expression into corresponding proteins.
Detailed Structure of Eukaryotic mRNA
1. 5' Cap Structure:
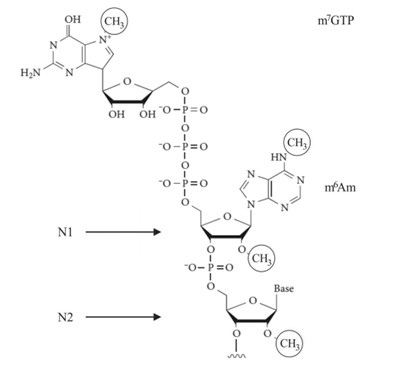 Cap structure of eukaryotic mRNA. (Jia, Longfei, et al., 2021)
Cap structure of eukaryotic mRNA. (Jia, Longfei, et al., 2021)
1. 5' Untranslated Region (5' UTR):
2. Open Reading Frame (ORF):
3. 3' Untranslated Region (3' UTR):
4. Poly-A Tail:
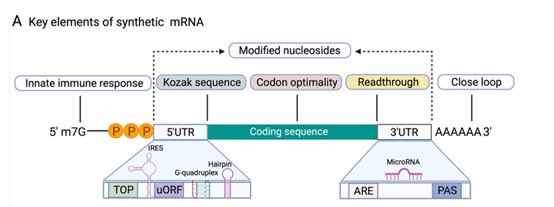 Schematic representation of a synthetic mRNA. (Jia, Longfei, et al., 2021)
Schematic representation of a synthetic mRNA. (Jia, Longfei, et al., 2021)
Role and Differences between Prokaryotic and Eukaryotic mRNA
mRNA plays a central role in gene expression in both prokaryotic and eukaryotic organisms, though structural and post-transcriptional modifications differ significantly between the two.
Key Differences between Prokaryotic and Eukaryotic mRNA:
| Feature | Prokaryotic mRNA | Eukaryotic mRNA |
|---|---|---|
| 5' Cap Structure | Absent | Present (7-methylguanosine cap, m7Gppp) |
| Poly-A Tail | Absent (may have shorter tails under certain conditions) | Present (typically 80-250 adenine residues) |
| Untranslated Regions (UTRs) | Short, generally not involved in regulation | Present (includes 5'UTR and 3'UTR, involved in regulation) |
| Introns and Exons | No introns (only exons) | Contains introns (non-coding) and exons (coding), requiring splicing |
| Post-transcriptional Processing | Minimal processing (no splicing or capping) | Extensive modifications (capping, splicing, and polyadenylation) |
| Stability | Relatively short-lived (minutes to hours) | More stable, regulated by 5' cap and 3' poly-A tail |
| Timing of Translation | Can initiate immediately after transcription | Translation begins only after mRNA processing and export to the cytoplasm |
| Localization of Transcription and Translation | Occurs in the cytoplasm (no nucleus) | Transcription in the nucleus, translation in the cytoplasm |
| Interaction with Ribosomes | Directly binds ribosomes post-transcription for translation | Requires cytoplasmic export and 5' cap-ribosome interaction for translation |
This distinction underscores the intricate regulatory mechanisms and structural complexities that differentiate mRNA functionalities in prokaryotes and eukaryotes, reflecting evolutionary adaptations in gene expression control.
mRNA-Seq stands as a transformative high-throughput technique, meticulously deciphering cell-specific mRNA sequences to unravel the complexities of gene expression and transcriptomics. This methodology offers profound insights across several critical dimensions:
Gene Expression Quantification: mRNA-Seq facilitates the accurate measurement of gene expression levels across a diverse array of samples, thus enabling the identification of genes that are differentially regulated under specific biological conditions. A valuable application includes contrasting the mRNA expression profiles of tumor versus normal cells to discern genes implicated in oncogenesis.
Transcript Variants and Isoforms: Beyond merely quantifying gene expression, mRNA-Seq is adept at delineating various transcript variants produced through alternative splicing, each potentially encoding functionally distinct protein isoforms. Such analysis underscores the technique's importance in exploring transcriptomic complexity and functional diversity.
Splicing and Post-Transcriptional Dynamics: The technology excels in characterizing splicing patterns and post-transcriptional modifications, such as methylation, which can profoundly influence gene expression. Identifying aberrant splicing patterns, often associated with genetic disorders, exemplifies the utility of mRNA-Seq in elucidating novel disease mechanisms.
Identification of Novel Genes and Transcripts: mRNA-Seq plays a pivotal role in the discovery of previously unannotated genes and novel transcripts, particularly in under-explored species or cell types. This enhances our genomic understanding, offering new insights into biological roles and disease pathways.
Applications of mRNA-Seq
The adaptability of mRNA-Seq finds relevance across multiple research fields:
Gene Expression Analysis: By assessing expression changes under diverse conditions, mRNA-Seq informs on gene regulatory mechanisms. It is particularly effective in disease research, where it helps pinpoint expression shifts pertinent to pathologies and identifies potential therapeutic targets.
Cancer Genomics: Within oncology, mRNA-Seq is instrumental in highlighting differences in gene expression between malignant and normal tissues. This approach aids in discovering genes crucial to tumor development and progression and identifies key biomarkers and therapeutic targets.
Precision Medicine: mRNA-Seq underpins the advances in personalized medicine, where individual mRNA expression profiles support the development of targeted therapeutic strategies—critical in cancer treatment for optimizing drug selection and therapeutic regimens.
Splicing and Transcriptomics Research: The technique is vital for investigating splicing patterns, especially given the association of many diseases, particularly genetic ones, with splicing irregularities. mRNA-Seq provides essential insights for early diagnostic and therapeutic innovations.
In conclusion, mRNA-Seq offers comprehensive insight into gene expression dynamics, transcript isoforms, splicing events, and post-transcriptional modifications. This makes it an indispensable tool in basic research, the study of disease mechanisms, personalized therapeutic strategies, and drug development. Leveraging mRNA-Seq enriches our comprehension of gene regulatory networks, laying a robust scientific foundation for breakthroughs in disease diagnosis, treatment, and pharmaceutical progress.
RNA sequencing (RNA-Seq) and mRNA-Seq are two prevalent genomics techniques used to analyze cellular transcriptomes, each with distinct areas of focus. The following delineates the principal differences between these methodologies:
| Features | Total RNA-Seq (RNA-Seq) | mRNA-Seq |
|---|---|---|
| Sequencing Target | Encompasses all RNA types (both coding and non-coding) such as mRNA, rRNA, tRNA, miRNA, etc. | Primarily targets coding RNA, specifically mRNA, often excluding other RNA types. |
| Sample Preparation Methods | Involves rRNA depletion followed by enrichment or selection of various RNA types. | Employs poly(A) enrichment techniques to specifically isolate mRNA, effectively removing rRNA. |
| Scope of Application | Offers comprehensive transcriptome data, including analysis of both coding and non-coding RNA. | Focused on gene expression analysis, ideally suited for studies of mRNA and its expression variations. |
| Sensitivity of Sequencing Data | High sensitivity for non-coding RNA (such as lncRNA and miRNA). | More focused on mRNA, providing deeper insights into coding regions. |
| Sample Requirement | Demands larger sample quantities due to the sequencing of all RNA types, resulting in substantial data output. | Requires smaller sample amounts and achieves higher sequencing depth due to mRNA enrichment. |
| Cost | Higher costs due to extensive data requirements and computational resources. | Lower costs, as it is confined to sequencing enriched mRNA, generating less data. |
| Analytical Outcomes | Facilitates analysis of all transcripts, both coding and non-coding, suitable for discovering novel transcripts. | Primarily analyzes coding RNA, invaluable for gene expression studies, particularly in disease research. |
| Appropriate Scenarios | Suitable for understanding complete transcriptome details, including non-coding RNA and post-transcriptional modifications. | Primarily addresses gene expression and transcript variations, notably useful in biomarker research. |
Detailed Comparison:
Target RNA Types:
Sample Preparation and Enrichment Methods:
Applicable Research Areas:
Cost and Data Volume:
Choosing Between mRNA-Seq and Total RNA-Seq:
The decision to use total RNA-Seq or mRNA-Seq should align with the research objectives and experimental constraints:
Both methodologies offer distinct advantages and limitations, necessitating a deliberate evaluation of sample type, research goals, financial resources, and desired depth of analysis prior to execution.
References:


