Precise annotation of clusters in Seurat plays a critical role in extracting valuable insights from single-cell RNA sequencing (scRNA-seq) datasets. By associating computationally detected clusters with biological relevance, researchers can better understand cellular heterogeneity and functionality. This guide offers a comprehensive step-by-step overview of methods, tools, and strategies for effective cluster annotation, aiming to achieve reliable and high-quality outcomes in scRNA-seq analyses.
Introduction to Cluster Annotation in Seurat
Seurat provides a versatile suite of tools commonly utilized for scRNA-seq data analysis. By enabling the grouping of cells according to gene expression profiles, it has significantly advanced the investigation of cellular populations. Nevertheless, clustering alone is insufficient; precise annotation is essential to interpret computational results in a biologically meaningful way. This process connects mathematical frameworks with biological contexts, helping researchers gain deeper insights into cellular diversity and roles.
Understanding Clustering in Seurat
What Is Clustering in Seurat?
Clustering in Seurat involves grouping cells into distinct populations based on their transcriptional profiles. This grouping is typically visualized using dimensionality reduction techniques like UMAP or t-SNE, which plot high-dimensional data in a two-dimensional space. Clusters represent discrete groups of cells that often correspond to specific cell types or functional states.
Why Is Annotation Important?
Annotation gives biological meaning to these computational clusters, ensuring that researchers can derive actionable insights from their data.
- Biological Relevance: Links computationally derived clusters to real-world cell types or states.
- Enhanced Interpretation: Clarifies the narrative of scRNA-seq data, making results more accessible and impactful.
- Downstream Analysis: Enables further studies, such as pathway enrichment and functional analysis.
Without accurate annotation, the biological utility of scRNA-seq analysis is diminished.
Methods for Cluster Annotation
Seurat provides flexibility in cluster annotation through manual, automated, and integrated approaches.
1. Manual Annotation
Manual annotation relies on prior knowledge of marker genes. By comparing the differentially expressed genes (DEGs) within each cluster against established markers, researchers can assign cell-type labels.
- Advantages: Allows expert interpretation and refinement.
- Challenges: Requires domain knowledge and can be time-consuming.
For instance, a cluster with high expression of CD3D and CD8A may be annotated as cytotoxic T cells. This approach is often used in studies where researchers have specific hypotheses about the cell types present. In one study, researchers manually annotated clusters from a scRNA-seq dataset of human peripheral blood mononuclear cells (PBMCs) by identifying clusters expressing known lymphocyte markers such as CD19 for B cells and CD3D for T cells, thus confirming their identities through literature-supported marker gene expression profiles(Zhao, J,et.al,2020).
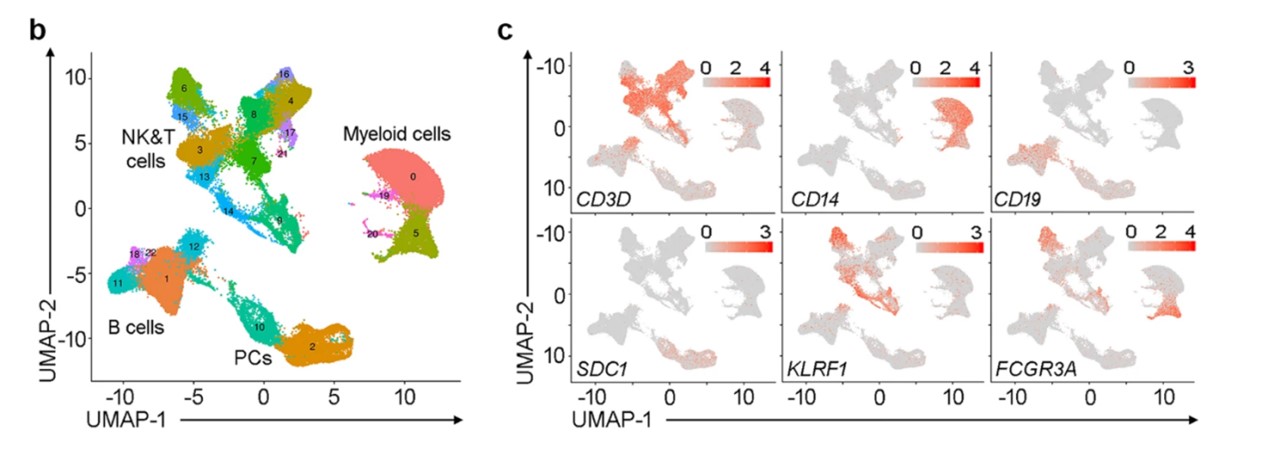 Figure1.UMAP plot of the immune cells(Zhao, J,et.al,2020).
Figure1.UMAP plot of the immune cells(Zhao, J,et.al,2020).
2. Automated Annotation
There are many software and methods for single-cell annotation, and as early as 2021, there was an article summarizing and comparing the advantages and disadvantages of different single-cell annotation software(Xie, B,et.al,2021).
Principle of automated Annotation
The principle of automatic cell type annotation leverages public single-cell RNA sequencing (scRNA-seq) data resources and algorithms to directly predict cell types without requiring manual annotation. It primarily includes three approaches: eager learning, which relies on classifiers; lazy learning, based on similarity to neighboring cells; and marker learning, which uses marker genes and scoring functions. These methods are trained on large-scale datasets and employ specific algorithms or scoring mechanisms to assign cell types in unknown data rapidly and accurately. This significantly improves analytical efficiency, making it suitable for large datasets and repeated analyses, while reducing dependence on domain expertise.
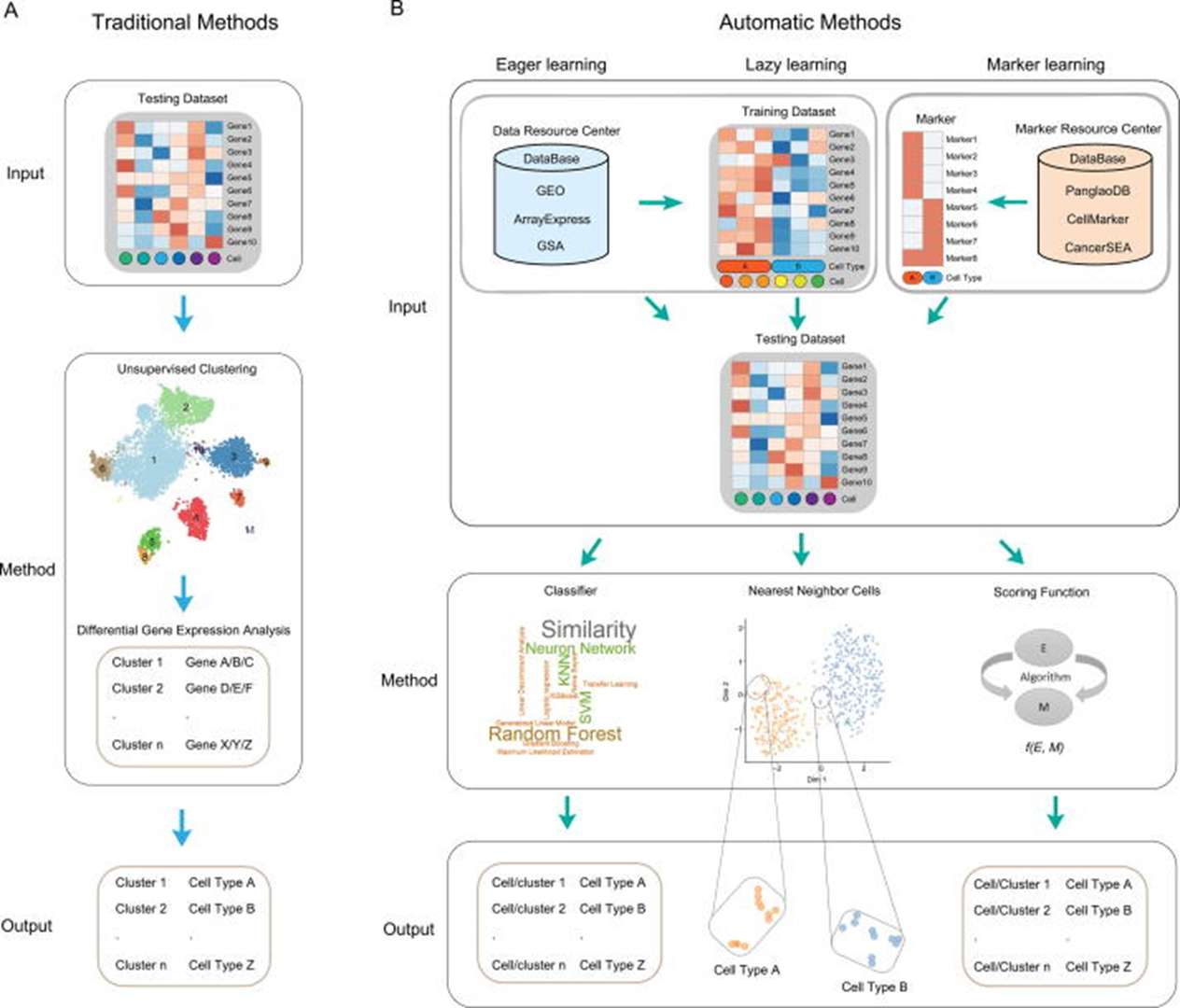 Figure2.Workflow of the traditional and automatic cell-type identification methods.(Xie, B,et.al,2021).
Figure2.Workflow of the traditional and automatic cell-type identification methods.(Xie, B,et.al,2021).
Seurat automated Annotation method
This method was first published in Nature Biotechnology (Butler, A.,et.al,2018). The researchers initially employed Canonical Correlation Analysis (CCA) to correct batch effects caused by non-biological factors across different samples. Given that the study was published relatively early, CCA may carry the risk of over-correction and can be time-intensive when integrating large datasets. Therefore, in practical applications, more advanced tools, such as Harmony or other integration methods, can be considered to construct reference datasets. Subsequently, the researchers identified single-cell types and their UMAP (Uniform Manifold Approximation and Projection) coordinates in the validation dataset through cell type label comparison and projection. In essence, the core of this method lies in leveraging known datasets to annotate unknown datasets and mapping the UMAP information of cells from the unknown dataset to the known dataset, ensuring that the same cell types from both datasets occupy approximately the same positions in the UMAP plot.
The reference data set on the left has basically eliminated the batch effect of different sequencing methods after CCA merger, and the different cell types on the right are clearly distinguished.
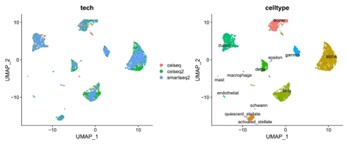 Figure3.CCA for data integrated and cell type prediction.
Figure3.CCA for data integrated and cell type prediction.
Using Marker Genes for Annotation
Marker genes are pivotal in cluster annotation, serving as identifiers for specific cell types.
Identifying Marker Genes
Seurat's FindAllMarkers() function identifies DEGs for each cluster. These genes are compared against known markers to assign biological identities. For example:
This function outputs a ranked list of genes associated with each cluster.
Common Marker Genes
| Cell Type |
Marker Genes |
| T Cells |
CD3D, CD4, CD8A |
| B Cells |
MS4A1 |
| Monocytes |
LYZ |
| NK Cells |
GNLY, NKG7 |
| Dendritic Cells |
FCER1A, CLEC10A |
Visualizing Annotated Clusters
Visualization is crucial for interpreting and communicating single-cell RNA sequencing (scRNA-seq) results. Seurat supports various visualization techniques to display annotated clusters effectively, allowing researchers to gain insights into complex datasets.
- UMAP: UMAP is a versatile tool that excels in preserving both local and global structures of the data. For instance, in a study analyzing immune cell populations, researchers applied UMAP to visualize clusters of T cells, B cells, and monocytes, which helped them understand the relationships between these immune cell types more clearly. The resulting UMAP plot revealed distinct clusters corresponding to different cell states, aiding in the identification of novel immune subtypes.
 Figure4.UMAP for reference annotations and query transferred labels.
Figure4.UMAP for reference annotations and query transferred labels.
- t-SNE: t-SNE is particularly useful for exploring local similarities among clusters. In a project examining tumor microenvironments, t-SNE was employed to differentiate between various tumor-infiltrating lymphocyte populations. The t-SNE plot highlighted closely related clusters, allowing researchers to identify specific immune responses associated with different tumor types. However, while t-SNE effectively visualizes local structures, it may sometimes obscure larger patterns present in the data(Kobak,et.al,2019).
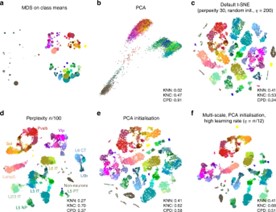 Figure5.t-SNE for cluster assignments(Kobak,et.al,2019).
Figure5.t-SNE for cluster assignments(Kobak,et.al,2019).
- Feature Plots: Feature plots are another powerful visualization tool within Seurat that allows researchers to highlight the expression of specific marker genes across clusters. For example, a feature plot displaying the expression of CD4 and CD8 markers can help distinguish between helper T cells and cytotoxic T cells within a cluster. This visualization technique is particularly beneficial for validating cluster identities based on known biological markers (Zhao, J,et.al,2020).
- Dot Plots: Dot plots summarize marker gene expression across clusters by displaying the average expression levels and the percentage of cells expressing each gene. In a study focusing on neuronal subtypes, dot plots were used to compare the expression of neurotransmitter receptors across different neuronal populations. This visualization provided a clear overview of how receptor expression varied among clusters, facilitating comparisons and biological interpretations.
- Heatmaps: Heatmaps provide detailed insights into gene expression patterns across multiple clusters simultaneously. Researchers often use heatmaps to visualize the expression of DEGs identified during cluster analysis. For instance, in research investigating stem cell differentiation, heatmaps were employed to show changes in gene expression profiles as stem cells transitioned into differentiated states. This approach allowed for easy identification of key regulatory genes involved in the differentiation process.
Conclusion
Cluster annotation in Seurat is a cornerstone of single-cell RNA sequencing research, enabling the discovery of cellular diversity and function. By leveraging marker genes, advanced tools, and visualization techniques, researchers can unlock profound biological insights.
References:
- Zhao, J., Zhang, S., Liu, Y. et al. Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov 6, 22 (2020). https://doi.org/10.1038/s41421-020-0157-z
- Xie, B., Jiang, Q., Mora, A., & Li, X. (2021). Automatic cell type identification methods for single-cell RNA sequencing. Computational and structural biotechnology journal, 19, 5874–5887. https://doi.org/10.1016/j.csbj.2021.10.027
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E., & Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature biotechnology, 36(5), 411–420. https://doi.org/10.1038/nbt.4096
- https://satijalab.org/seurat/articles/integration_mapping.html
- Kobak, D., Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat Commun 10, 5416 (2019). https://doi.org/10.1038/s41467-019-13056-x

 Sample Submission Guidelines
Sample Submission Guidelines


