Because sequencing technologies generate DNA sequences in pieces, genome assembly is the process of reconstructing a complete genome from smaller sequences. This is an in principle a simple problem, but one that is essential for recovering genetic information from the most primitive microbial genomes all the way up to very complex plant and animal genomes. Modern assembly workflows are constructed around the premise that they must work with high-fidelity datasets to deal with, e.g., repetitive sequences, sequencing errors or variability in the genome.
Overall Process of Genome Assembly Steps
This is an iterative process, whereby the steps taken along the way lead to an improved genome in terms of quality and accuracy. They consist of data preprocessing, assembling, scaffolding, polishing and validation. It isn't trivial to reconstruct the genome from these fragments because of things like differing expression, errors, and repetitive elements-all these are common in the genomic data, and so the above processes guarantee that the genome we construct is correct and complete. The stages demonstrate a significant aspect of contiguating dis-contiguous assemblies into a single biologically meaningful genome.
Service you may interested in
Resource
Data Preprocessing
Before detailed analysis, raw sequencing data requires preparation for genome assembly. This step removes low-quality reads, contaminants and sequencing artifacts in order to maintain the accuracy of the assembly. You are recommended to preprocess your data properly, else you may run through errors or biases that can propagate the entire assembly pipeline and greatly affect the final results.
QC Quality control: QCQuality control is done using FastQC on the sequencing reads. Findings include adapter contamination, base composition biases and low-quality areas. FastQC reports provide the detail that allows iterative data-cleaning steps to be applied to retain high-quality reads for assembly. FastQC provides graphical summaries of your data which will tell you, in one glance, if you have a problem with your data.
Cleaning and Pre-filtering: The use of tools (Trimmomatic or Cutadapt) to Clean reads by removing adapters, low-quality bases, and reads that are too short. If you have contamination from the adapter, it would not give an accurate reconstruction of contigs, and low-quality bases may even lead to errors in assembly. But in the downstream that's precisely where recently trimmed, high quality data is an excellent starting point for accurate assembly. Moreover, these tools include an option to define trimming thresholds, allowing researchers to use the trimming features in a manner that fits the specificity of their preprocessing pipielines.
Error Correction: Long-read sequencing platforms (PacBio, Nanopore) usually produce larger errant reads due to limitations with the underlying sequencing technology. Tools for read error correction (e.g. Racon and Canu) correct the reads by long self-alignment (the reads align to each other) or by aligning the reads to high-quality short reads, both of which substantially improve the read quality for the assembly. The most critical in this process is in organism with larger genome as leaky error in long read, if left uncorrected, result in gross error which will skew the read multiple time during mapping.
Assembly
During the assembly phase, reads are assembled into longer contiguous sequences, or contigs. It is the most crucial step in genome assembly and demands algorithms capable of coping with this complicated dataset whilst preserving the structural integrity of the genome. Choosing the assembly strategy is one of the critical decisions in any genome assembly pipeline, because it relies on the sequencing platform, genome size, and the aims of the project.
De Novo Assembly (Reference free): Build genomes from scratch without reference. De novo assembly is required for novel organisms or organisms sufficiently distant from the available reference genomes. Some examples are SPAdes (for short reads) and Flye (for long reads) for de novo assembly. These allow reconstruction of contiguous sequences using graph-based algorithms to avoid pitfalls of repetitive regions and sequencing errors. De novo assembly is often the method of choice for characterization of microbial diversity or discovery of new species.
Tools-Aided: Existing reference genome provides scaffold. The reference aids in assembly based on: Reads mapped to the reference. This Scheme is computationally less demanding and precise for closely related species. Data will then be processed using tools such as BWA and Bowtie2 for alignment, followed by SAMtools for cleanup prior to assembly. Studies oriented towards resequencing can use reference-guided assembly to quickly determine differences relative to a reference. Although this is a relatively fast approach and has a good trade-off between speed and accuracy, it might have difficulties dealing with novel sequences not found in the reference.
Hybrid Assembly: The short- and long-reads can be combined to obtain the accuracy of short reads and structural resolution of long reads. We would like to make specific mention of programs such as MaSuRCA and Unicycler that are used for hybrid assembly and provide better assembly of complex regions of genomes. Hybrid assembly relies on data from multiple platforms to yield highly contiguous assemblies that conform to the organization of the genome. This is especially valuable to also resolve repeats and structural variants that are difficult to assemble with any single data type.
Scaffolding and interleaved fill-gap
Scaffolding connects contigs into bigger structures using additional information, e.g. mate-pair reads, long reads or Hi-C data. This is a crucial step in assembly of chromosomal arrangements and larger genomes. Scaffolding designs take joining of the assemblies, by filling within to investigate between neighbouring contigs, which progressively builds genome representation. Proper scaffolding is needed for biologically meaningful assemblies that reflect the genome architecture.
Scaffolding tools: Scaffolding tools are, for instance LINKS and SSPACE that order and orient contigs using informations from paired-end and mate-pair reads. Hi-C-derived methods such as 3D-DNA produce scaffolds at the chromosome level, dependent on the ability to capture spatial interactions between genomic loci. These methods have revolutionized large-genome assembly and enabled researchers to produce assemblies that span entire chromosomes. Machine learning-based scaffolding algorithms have been developed recently to improve contig placement and orientation.
Analysis Tools: Hardware and software bioinformatics tools used to evaluate if contigs assembled correctly. These tools are capable of increasing the completeness of assembly by leveraging extra sequencing data to fill in the sequence gaps, further extending their utility. For example, and algorithm PBJelly uses long reads to fill in the gaps by properly placing repetitive or structurally complex regions in the assembly. At the core of the assembly process, gap-filling is important for producing high-quality genomes for genomic analyses that rely on high resolution.
Polishing
Polishing ensures high base-level accuracy of the assembled genome. This phase has a much more prominent role in assemblies generated from long-read platforms, with relatively high raw error, than in any of the other platforms. Refining base calls and correcting residual errors during this polishing step can significantly improve the usability of genomes and their subsequent uses, e.g., for gene prediction and variant calling.
Polishing tools: Nanopolish and Medaka for Oxford Nanopore data, Arrow and Racon for PacBio assemblies Anomalies are fixed with help of residual base error fixing tools and align criteria increases agreement by re-aligning reads back to the assembly and checking for differences. Polishing algorithms are growing into more sophisticated varieties, using models able to catch minor errors that standard correction generally overlooks.
Iterative polishing: For very large or error-prone datasets, getting a highly complete genome can require multiple rounds of polishing. This enables iterative polishing that corrects all but the most subtle of errors and produces assemblies that are suitable for high precision tasks like variant calling and gene prediction. Adopting iterative approaches allows use of jobs that 148 cover complex regions for consensus-based pipelines, including repeated element or elevated GC area.
Decomposing and estimating the quality
Validation involves checking the quality, completeness, and correctness of the assembled genome. Doing this has the added benefit of ensuring your assembly meets your project-level quality control benchmarks, which will be useful for downstream analyses. Running a validation process not only builds confidence in the assembly but also highlights elements within the assembly that require improvements. This is the final threshold before the sequenced genome can be used in biological research.
Assembly statistics: N50 (locality-defined), L50, and genome sizes. These statistics provide a quantitative measure of the assembly performance, but also insight into the fragmentation and completeness of the assembly. Large N50 values mean assemblies are repeated frequently and L50 values give an estimate of how that assembly is distributed.
Completeness: Using tools like BUSCO to look for conserved genes in your assembly. A BUSCO score > 90 denotes that a significant proportion of expected genomics content is present in the assembly, designating it a great resource for functional studies. Completeness assessments enable the identification of poorly-sampled or missing areas that could benefit from closer attention.
Viewing reply thread
Genome assembly is a complex problem characterized by multiple biological and technical obstacles. This is magnified in large, complex genomes, where repetitive sequences, heterozygosity, and sequencing errors complicate the reconstruction task. Addressing this helps enable the generation of better-quality assemblies that more accurately reflect the genome under investigation.
Highly repetitive regions: These areas can lead to fragmented assemblies or misassemble. Long-read sequencing technology like PacBio and Nanopore have enough resolving power to span repeats. Hybrid assembly combining long and short reads also performs well in resolving repetitive regions. There is another class of assembly algorithms, however, that are based on processing the read fragment graphs and focus on handling sequence repeats.
Heterozygosity: Variation between homologous chromosomes results in particular challenges for assembly (especially in diploid and polyploid organisms). Haplotype-resolving assemblers and phasing tools handle this by separating homologous sequences of haplotypes before accurately reconstructing them. Cross-species modelling may be especially relevant for exploring admixture in populations or for elucidating evolutionary history in polyploid species.
Data Size: A large dataset, can be memory expensive and requires high processing power. Such demands are being accommodated more and more, which are cloud-based platforms and parallelized algorithms. These approaches help genomics researchers manage the computational cost of the people building large genomes. Also, light assemblies brought genome assembly within reach of labs with spare computing power.
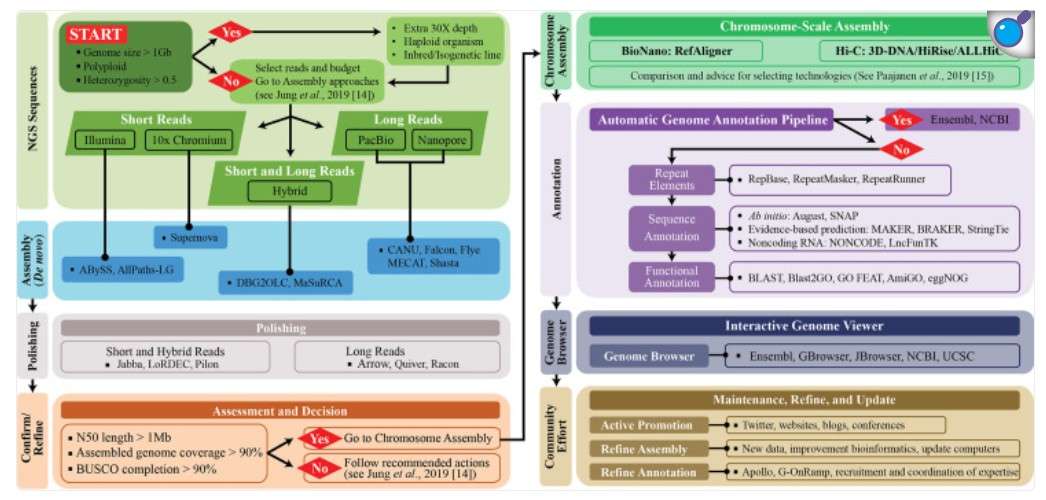 Recommended flowchart for genome assembly and annotation (Jung, H. et al 2024).
Recommended flowchart for genome assembly and annotation (Jung, H. et al 2024).
Case study: Human Genome Assembly
Background
The human genome - with its ~3 billion base pairs and large repetitive regions - required innovative strategies and extensive resources for assembly. This project set the stage that would fuel modern genomics and transform our understanding of human biology and disease. Its success demonstrated that sequencing and assembling complex genomes was achievable - and was seen as an important precursor for other advances in the field.
Methods
Hierarchical assembly with shotgun sequencing: The genome was divided into large chunks, and then shotgun sequenced separately using bacterial artificial chromosome (BAC) technology. In a second strategy for reducing this complexity and increasing assembly accuracy, Smits et al. smile by zeroing in on smaller, more controllable parts of the genome. This hierarchical strategy provided a solution for tackling the problems posed by high-complexity and repetitive genomics de novo.
Scaffolding & Finishing: contigs were linked into longer sequences using high-resolution maps and computational scaffolding. Supplementary sequencing data and manual curation were used to fill in gaps within a gapless genome. Such the advanced genomic assembly, came with these advanced computational and manual methods, resulted in the most complete and the highest quality assembled genome.
Results
A high-quality reference genome generated by this project has now been enhanced through the use of superior sequencing and assembly technologies. The human reference genome remains an essential framework for the thousands of biomedical studies that explore genetic variation, disease mechanisms, and evolutionary processes. It also facilitated invention of new assembly techniques and tools that propelled genomics. The success of the Human Genome project has led to the establishment of numerous international initiatives directed towards sequencing other complex genomes.
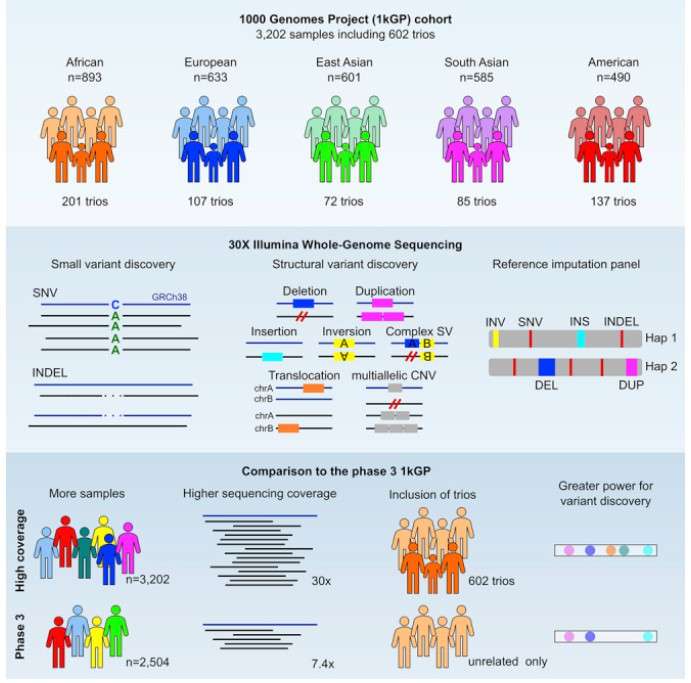 Genome assembly steps (Byrska-Bishop, M. et al 2024).
Genome assembly steps (Byrska-Bishop, M. et al 2024).
Conclusion
Genome assembly - the reconstruction of whole genomes - is a pivotal genomic tool that is a prerequisite to the biology underpins biological exploration. It is this combination of high quality assemblies produced by a structured pipeline, and high throughput sequencers that allows these applications to be made. Research on genome assembly will be central to the understanding of this complexity, and as the field moves ahead, genome assembly development will always close to the cutting edge in addressing critical issues in life sciences. Genome assembly will probably become increasingly precise, faster and more accessible than ever before with each new wave of technical progress and will at the same time find newer arenas of application in science and medicine.
References:
- Jung, H., Ventura, T., Chung, J. S. et al (2020). Twelve quick steps for genome assembly and annotation in the classroom. PLoS computational biology, 16(11), e1008325. https://doi.org/10.1371/journal.pcbi.1008325
- Byrska-Bishop, M., Evani, U. S., Zhao, X. et al (2022). High-coverage whole-genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. Cell, 185(18), 3426–3440.e19. https://doi.org/10.1016/j.cell.2022.08.004

 Sample Submission Guidelines
Sample Submission Guidelines


