
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

As microbiological research advances, the importance of species-level identification in microbial communities becomes increasingly evident. Accurate identification of key microorganisms at the species level is critical for subsequent experimental validation and mechanistic discovery. Traditional second-generation sequencing (SGS) technologies, constrained by short read lengths, typically capture only 1-2 variable regions of the 16S rRNA gene (e.g., the V3-V4 region). This limitation hampers the ability to fully and accurately identify species within microbial communities, rendering species-level analyses incomplete. In contrast, full-length 16S rRNA sequencing technology captures all nine variable regions (V1-V9) in a single read, dramatically enhancing the precision of species-level annotation. Consequently, this technology offers substantial advantages for microbial community research, particularly in fields such as gut, respiratory, reproductive, environmental, livestock, and aquaculture microbiomes, where it has been widely applied.
The figure below compares the reconstruction of a mock microbial community using both SGS and full-length diversity sequencing. At the species level, the SGS results display several unannotated taxa and a lack of coherence, while the full-length sequencing results accurately reconstruct all the species present in the mock community, with consistent and precise annotations.
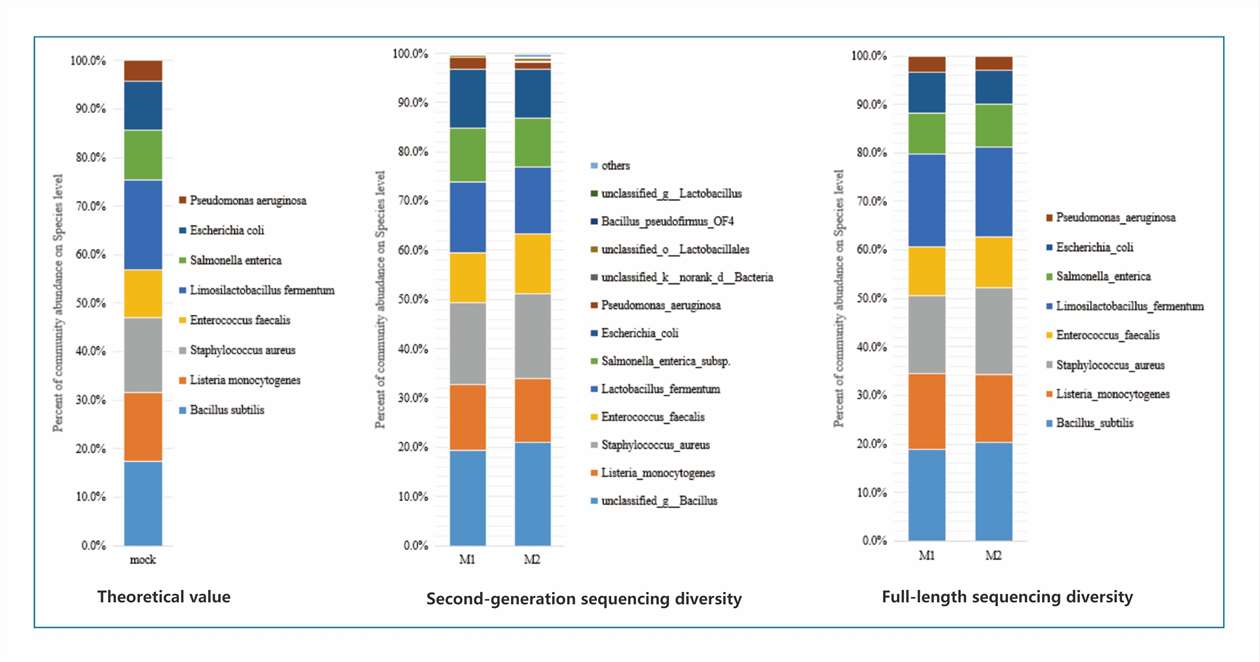 Figure 1. Reconstruction of the mock community using second-generation diversity and full-length diversity, respectively.
Figure 1. Reconstruction of the mock community using second-generation diversity and full-length diversity, respectively.
The table below presents the species annotation rates for common samples using full-length 16S rRNA sequencing. Notably, the species-level annotation rates for full-length sequences are highly satisfactory. For fecal and gut content samples, the species-level annotation rate reaches 90%, while for more complex environmental samples, such as soil and water, the rate still reaches 75%. This indicates that the vast majority of operational taxonomic units (OTUs) or sequences can be accurately annotated to specific species, providing significant value in the rapid identification of key microorganisms in samples.
| Sample Type | Genus-Level Average Annotation Rate (OTU) | Species-Level Average Annotation Rate (OTU) | Genus-Level Average Annotation Rate (Reads) | Species-Level Average Annotation Rate (Reads) |
| Feces | 85% | 85% | 90% | 90% |
| Gut Content | 85% | 85% | 90% | 90% |
| Saliva/Sputum | 90% | 87% | 93% | 87% |
| Nasal/Oral Swab | 88% | 85% | 95% | 89% |
| Vaginal Swab | 85% | 86% | 90% | 85% |
| Skin Swab | 82% | 80% | 90% | 88% |
| Soil | 78% | 73% | 82% | 75% |
| Water | 72% | 70% | 82% | 75% |
| Rumen | 83% | 80% | 88% | 85% |
| Sludge | 70% | 70% | 75% | 72% |
| Fermentation | 93% | 82% | 95% | 90% |
The species- and genus-level annotation rates, based on both OTU and read-level data, clearly demonstrate the superior performance of full-length 16S rRNA diversity sequencing in microbial community research. This advantage is particularly evident in clinical, agricultural, and environmental studies, where comprehensive and accurate microbial profiling is essential.
The practical results outlined above underscore the substantial advantage of full-length 16S rRNA diversity sequencing in identifying species-level organisms. From a technical perspective, several factors contribute to this enhanced performance.
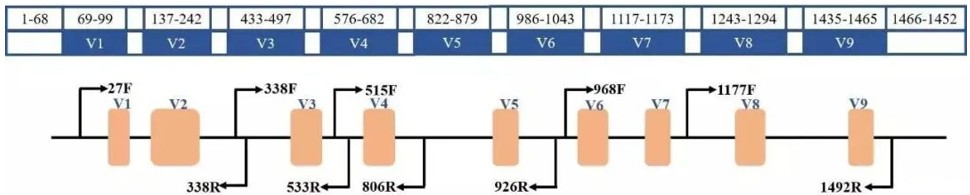 Figure 2. Structure of 16s
Figure 2. Structure of 16s
Second-generation sequencing technologies typically generate shorter read lengths, covering only one to two variable regions of the 16S rRNA gene. In contrast, the third-generation PacBio platform produces long-read sequences with an average length ranging from 8 to 15 kilobase pairs (kb), enabling comprehensive coverage of all nine variable regions.
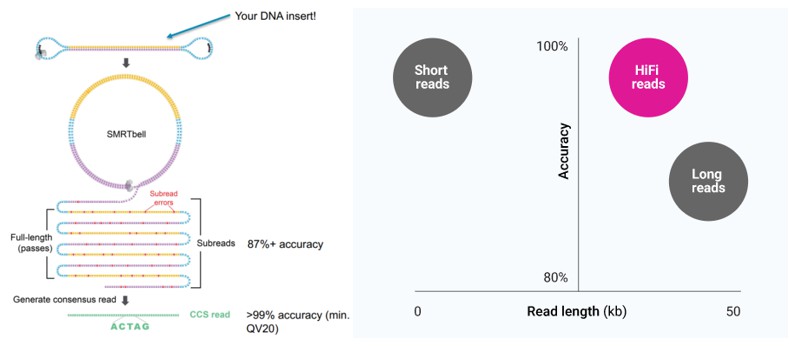 Figure 3. Sequencing Workflow Diagram (Left) and PacBio Website Demonstration of Sequencing Accuracy (Right)
Figure 3. Sequencing Workflow Diagram (Left) and PacBio Website Demonstration of Sequencing Accuracy (Right)
1. Precise species identification
2. Accurate reconstruction of microbial community composition
In 2019, Johnson et al. conducted a comprehensive reassessment of the taxonomic potential of the 16S rRNA gene at the species and strain levels, using both bioinformatic analysis and sequencing experiments. Their findings indicated that targeting short-read sequencing platforms with variable regions of the 16S gene does not provide the taxonomic resolution achieved by sequencing the entire gene (~1500 bp).
This conclusion highlights a significant limitation in short-read sequencing methodologies, where only partial regions of the gene are captured. In contrast, full-length 16S sequencing—such as that enabled by PacBio's continuous long-read sequencing—enables a much higher resolution of species and strain-level differentiation, ensuring the accuracy of microbial community profiling.
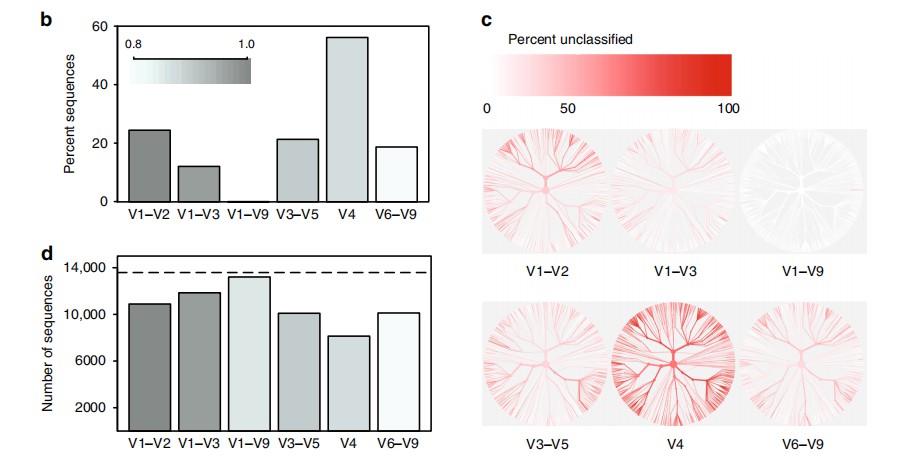
The authors observed that certain hypervariable regions of the 16S rRNA gene were able to differentiate microbial species to varying degrees (Figure B). However, full-length 16S rRNA sequencing demonstrated the capacity to accurately resolve all sequences at the species level. A comparison of different variable regions revealed biases in the bacterial taxonomic classification (Figure C).
The selection of variable regions significantly influenced the number of OTUs generated during clustering. When clustering sequences at 99% similarity, several variable regions failed to reproduce the actual number of taxa present in the microbial community (Figure D).
These findings emphasize the limitations of partial 16S sequencing approaches, particularly in environments where high taxonomic resolution is necessary. Full-length 16S sequencing, by covering all nine hypervariable regions, provides a more comprehensive and accurate representation of microbial diversity.
You may interested in
Learn More
Title: Bacterial Communities of Cassiopea in the Florida Keys Share Major Bacterial Taxa with Coral Microbiomes
Published in: bioRxiv
Tech: Full-Length 16S rRNA Diversity Analysis
In 2023, our research team published a preprint on bioRxiv utilizing PacBio Sequel technology to investigate the bacterial communities associated with Cassiopea medusae in the Florida Keys. The study aimed to uncover the dynamics of these microbial communities and their relationships with coral-associated bacteria.
Through comprehensive full-length 16S rRNA sequencing, we identified significant bacterial taxa prevalent in both Cassiopea and nearby coral microbiomes, such as Endozoicomonadaceae and Vibrionaceae. Our findings revealed distinct microbial compositions between the internal (gastrovascular cavity) and external (mucus) communities of the medusae, with the internal community exhibiting lower diversity compared to the external and environmental samples.
We also observed spatial variations in community composition across different sites, indicating that while Cassiopea does not possess a strict core microbiome, certain taxa consistently associate with these medusae. This highlights the ecological importance of these bacterial families and their potential role in the health of Cassiopea.
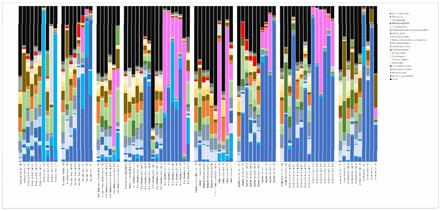 Order level bar plots grouped by location
Order level bar plots grouped by location
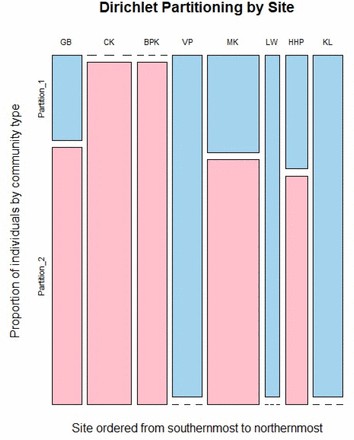 Mosaic plot of Dirichlet partition identity (community type) by site
Mosaic plot of Dirichlet partition identity (community type) by site
This study enhances our understanding of cnidarian microbiomes and their ecological significance, providing foundational data for future research on microbial dynamics in marine ecosystems. The implications extend to discussions on symbiotic relationships and ecosystem health, particularly in the context of changing environmental conditions.
Title: Microbial Metabolite Enhances Immunotherapy Efficacy by Modulating T Cell Stemness in Pan-Cancer
Journal: Cell
Omics Technologies: Full-length 16S rRNA Diversity, Metagenomics, Untargeted Metabolomics, Single Bacterial Genome, Single-Cell Transcriptomics
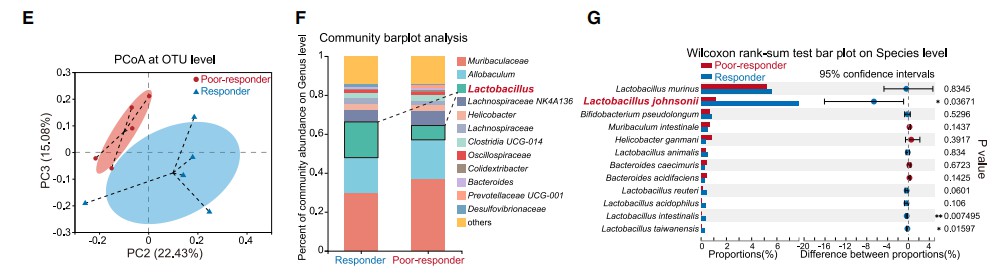
In March 2024, Cell published a groundbreaking study unveiling a novel anti-cancer mechanism mediated by gut microbiota. This study initially demonstrated, through fecal microbiota transplantation (FMT) experiments in mice, that the gut microbiome has a direct influence on the response to immune checkpoint blockade (ICB) therapy.
Using full-length 16S rRNA sequencing, the researchers compared the fecal microbiomes of mice exhibiting poor and strong responses to ICB therapy. The analysis at the genus level revealed a decrease in the abundance of Lactobacillus species in the poor-response group. At the species level, Lactobacillus johnsonii exhibited the most significant differential abundance between the two groups (as shown in the accompanying figure).
After identifying L. johnsonii as a key species, the researchers further investigated its functional role. Animal experiments demonstrated that oral administration of L. johnsonii enhanced the efficacy of anti-PD-1 therapy and increased the proportion of CD8+ T cell infiltration in tumors. In subsequent studies, employing metabolomics, bacterial genomics, and single-cell transcriptomics, the authors explored how L. johnsonii and other critical gut microbes modulate the immune response to cancer therapy.
The results confirmed that L. johnsonii enhances ICB efficacy by promoting the synthesis of indole-3-propionic acid (IPA) and boosting the activity of progenitor-exhausted T cells (Tpex). These findings offer new insights into how gut microbiota can be harnessed to improve cancer immunotherapy outcomes.
CD Genomics offers a complete suite of omics services, now with upgraded one-stop delivery standards. Our comprehensive product portfolio includes microbial diversity analysis, metagenomics, metatranscriptomics, microbial genomics, plant and animal resequencing, transcriptomics, prokaryotic transcriptomics, small RNA sequencing, lncRNA analysis, whole-transcriptome sequencing, single-cell sequencing, and spatial transcriptomics, among others.
References:


