
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

In recent years, numerous researchers have dedicated themselves to the in-depth exploration of microbial communities within the medical field, particularly focusing on the identification of key species at the species level. This identification is crucial for subsequent mechanistic studies and experimental validation. Although next-generation sequencing (NGS) technologies can annotate microbial communities at the species level, the short sequence lengths often undermine the accuracy of the results. Conversely, metagenomic analyses can achieve species-level resolution; however, the microbial data generated from samples with high host content—such as tissues and intestinal contents—are frequently limited.A pertinent question arises: is there a sequencing technology capable of addressing both the resolution of species identification and the influence of host-derived sequences?
The article titled Full-Length 16S rRNA Sequencing Enhances Species-Level Microbial Analysis has elucidated the significant advantages of CD Genomics in the analysis of full-length microbial diversity. As CD Genomics continues to research and optimize its techniques, a notable enhancement in species-level annotation rates has been observed, substantially facilitating the discovery of species within clinical and animal samples.
A review of recent literature concerning microbial communities in medical research indicates that full-length microbial diversity analysis has been widely applied across various fields, including studies on the gut, respiratory, and reproductive tracts. Consequently, this manuscript consolidates several research articles that employ full-length microbial diversity analysis within clinical cohort studies for the reader's reference.
Microbial Translocation from the Oral Cavity to Nasopharyngeal Carcinoma Patients
Published in: Nature Communications
Impact Factor: 14.7
Techniques Used: Next-Generation Sequencing, Full-Length 16S Microbial Diversity, Bacterial Genomics, Meta-Transcriptomics
Abstract
This study examined microbial translocation from the oral cavity to nasopharyngeal carcinoma (NPC) in a cohort of patients. A total of 165 untreated NPC patients and 138 non-malignant controls were included in the analysis. Paired nasopharyngeal swabs and saliva samples were collected for full-length microbial diversity sequencing and next-generation sequencing of 16S rRNA genes.
Results
Microbial Identification: In both the NPC and control groups, paired nasopharyngeal-oral samples revealed the identification of 13 microbial species that exhibited abnormal translocation from the oral cavity to the nasopharynx, with notable enrichment in NPC patients.
Validation of Microbes: Fusobacterium nucleatum and Prevotella intermedia were confirmed through culture-based methods and clonal strain identification.
Meta-Transcriptomic Evidence: Meta-transcriptomic analyses of nasopharyngeal biopsies corroborated the presence of these microbes within tumor tissue, suggesting an influence on the local microenvironment and cytokine responses.
Association with Epstein-Barr Virus: A significant correlation was observed between these microbes and Epstein-Barr virus (EBV) load within the nasopharynx, indicating an increased dose-response relationship.
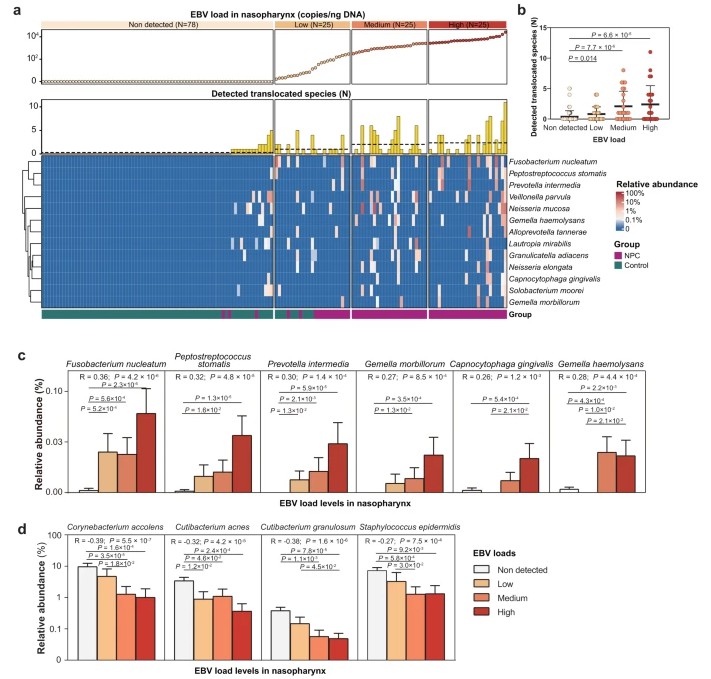 Fig. 1: The association between oral translocated microbes and EBV infection in nasopharynx.
Fig. 1: The association between oral translocated microbes and EBV infection in nasopharynx.
Conclusion: This study establishes that oral microbes can migrate to the nasopharynx and infiltrate tumors, thereby influencing the tumor microenvironment and correlating with EBV infection.
This research underscores the relevance of oral microbiota in the pathogenesis of nasopharyngeal carcinoma, warranting further investigation into their roles in oncogenesis and local immune modulation.
You may interested in
Learn More
Enrichment of Oral Microbiota in Early Cystic Precursors to Invasive Pancreatic Cancer
Published in: Gut
Impact Factor: 23
Technique: Full-Length 16S Microbial Diversity
Abstract
The present study investigates the potential microbial communities present within pancreatic cystic tumors (PCNs) in a cohort of surgical patients. A total of 105 patients with suspected pancreatic cystic tumors were included in the analysis, focusing on the microbiological characteristics of these lesions.
Results
Microbial Assessment: Significant elevations in bacterial DNA load and the inflammatory marker interleukin-1 beta (IL-1β) were observed in cyst fluid derived from intraductal papillary mucinous neoplasms (IPMNs) exhibiting high-grade dysplasia and carcinoma when compared to non-IPMN patients.
Specific Bacterial Enrichment: Within the cystic fluid of high-grade dysplastic IPMNs, oral bacterial taxa, including Fusobacterium nucleatum and Granulicatella adiacens, were concurrently detected and found to be enriched.
Clinical Correlations: The increase in bacterial DNA within cystic fluid was correlated with a history of invasive endoscopic procedures. This correlation was independent of proton pump inhibitor (PPI) and antibiotic usage.
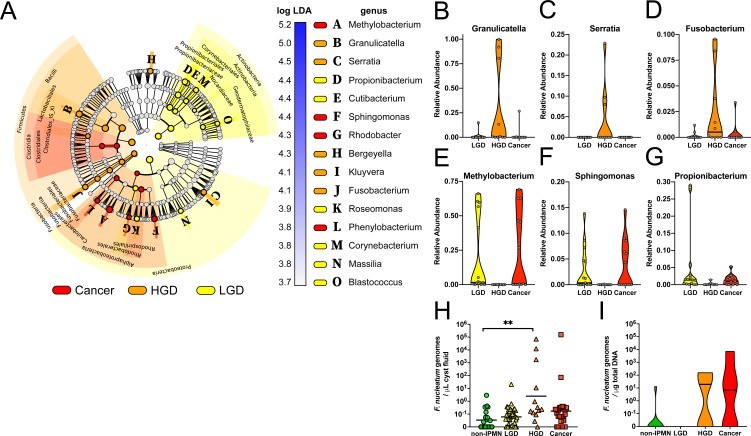 Figure 2: (A) Cladogram illustrating linear discriminant analysis effect size results that identified 15 differentially abundant features among the diagnostic classes (red=Cancer, orange=IPMN HGD, yellow=IPMN LGD). (B–G) Violin plots displaying the relative abundance distribution and median of selected bacterial genera by diagnostic group, with validation of Fusobacterium nucleatum genome quantification via TaqMan qPCR.
Figure 2: (A) Cladogram illustrating linear discriminant analysis effect size results that identified 15 differentially abundant features among the diagnostic classes (red=Cancer, orange=IPMN HGD, yellow=IPMN LGD). (B–G) Violin plots displaying the relative abundance distribution and median of selected bacterial genera by diagnostic group, with validation of Fusobacterium nucleatum genome quantification via TaqMan qPCR.
Conclusion
This research highlights the enrichment of oral microbiota in early cystic precursors to invasive pancreatic cancer, suggesting a potential link between microbial dysbiosis and pancreatic oncogenesis. Further studies are warranted to elucidate the implications of these findings for early detection and treatment strategies.
The Meconium Microbiota Shares More Features with the Amniotic Fluid Microbiota than the Maternal Fecal and Vaginal Microbiota
Published in: Gut Microbes
Impact Factor: 12.2
Technique: Full-Length 16S Microbial sequencing
Abstract
This study examines the microbial communities present in meconium and maternal samples, including amniotic fluid, feces, vaginal fluid, and saliva, in a cohort of 39 mother-infant pairs. The findings elucidate the similarities and differences in microbial composition across these samples.
Results
Sample Analysis: A total of 39 mother-infant pairs were analyzed, focusing on the microbiota present in meconium and various maternal samples.
Diversity Analysis: Both alpha and beta diversity analyses revealed distinct microbial community characteristics specific to each sample type.
Dominant Genera Identification: The predominant genera identified were as follows:
Amniotic Fluid: Lactobacillus and Campylobacter.
Vaginal Samples: Bacillus and Escherichia/Shigella.
Maternal Feces: Bacteroides and Faecalibacterium.
Saliva Samples: Streptococcus and Salmonella.
Microbial Origins: The meconium microbiota was found to originate from multiple maternal sites, with several operational taxonomic units (OTUs) shared across maternal samples. Notably, the amniotic fluid microbiota exhibited the greatest influence on the composition of the meconium microbiota.
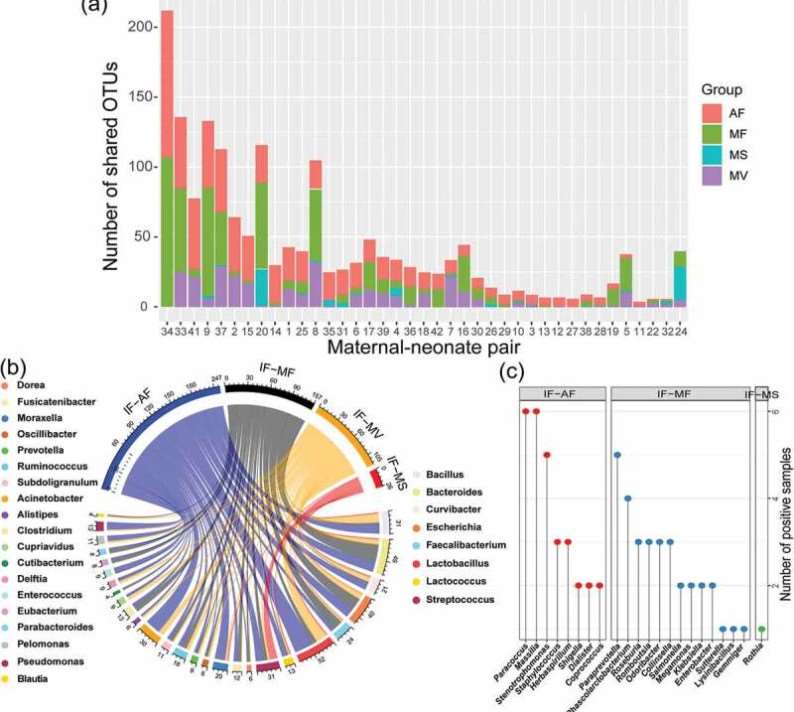 Figure 3: Prediction of the maternal origin of the meconium microbiota by dyad analysis.
Figure 3: Prediction of the maternal origin of the meconium microbiota by dyad analysis.
Conclusion
These findings suggest that the meconium microbiota bears a closer resemblance to the amniotic fluid microbiota than to the maternal fecal and vaginal microbiota, indicating a complex interplay of microbial communities during the perinatal period. Further research is warranted to explore the implications of these microbial interactions for neonatal health.
Probiotic-Directed Modulation of Gut Microbiota is Basal Microbiome Dependent
Published in: Gut Microbes
Impact Factor: 12.2
Technique: Full-Length 16S Microbial Diversity Analysis
Abstract
This study investigates the effects of Lactobacillus casei Zhang (LCZ) on the gut microbiota of 106 healthy adults from six regions in Asia. The modulation of gut microbiota by probiotics is shown to be dependent on the baseline composition of the microbiome, demonstrating regional variations.
Results
Study Cohort: A total of 106 healthy adults from six different Asian regions were enrolled to analyze the impact of LCZ on gut microbiota.
Microbiota Composition Dependence: The influence of LCZ on microbial composition was found to correlate closely with the participants' baseline gut microbiota, exhibiting significant regional differences.
Effects on Microbial Abundance: Consumption of LCZ resulted in an increase in the relative abundance of beneficial bacteria while inhibiting the growth of harmful bacteria. Additionally, changes in gut type were observed in certain participants, leading to an elevation in the abundance of lactic acid bacteria.
Metabolic Enhancements: LCZ was shown to enhance microbial phosphate metabolism, amino acid transport systems, and isoleucine biosynthesis, while simultaneously reducing lipopolysaccharide (LPS) biosynthesis.
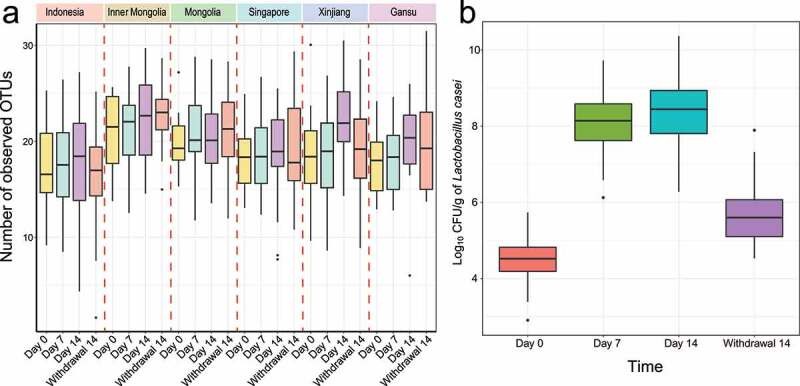 Figure 4. Effect of consumption of the probiotic LCZ on the gut LAB of participants from six regions.
Figure 4. Effect of consumption of the probiotic LCZ on the gut LAB of participants from six regions.
Conclusion
The findings indicate that the probiotic-directed modulation of gut microbiota is significantly influenced by the basal microbiome composition. This underscores the importance of personalized probiotic interventions based on individual microbial profiles for optimizing health outcomes. Further research is recommended to explore the implications of these findings in diverse populations.
High-Resolution Detection of Translocation of Oral Bacteria to the Gut
Published in: Journal of Dental Research
Impact Factor: 5.7
Technique: Full-Length 16S sequencing
Abstract
The aberrant enrichment of oral microbiota in the gastrointestinal tract represents a significant alteration in gut microbial equilibrium. It is hypothesized that these microorganisms may be translocated via saliva and food; however, evidence supporting oral-gut microbial transmission remains insufficient and warrants further investigation.
Results
Study Cohort: The study encompassed 144 participants from a community in xx Town, Japan, across various age groups. Saliva and fecal samples were collected for high-resolution full-length 16S microbial diversity sequencing.
Microbial Composition Differences: A marked distinction was observed in the bacterial composition between saliva and gut microbiota. Nevertheless, in 72.9% of participants, at least one amplicon sequence variant (ASV) was shared between the saliva and gut microbiota.
Proportional Sharing of ASVs: The shared ASVs constituted between 0.0% and 63.1% (median 0.14%) of the gut microbiota per participant, with notable representations of Streptococcus salivarius and Streptococcus parasanguinis. Elderly participants exhibited a significantly higher total relative abundance of these bacteria, correlating with increased dental plaque accumulation.
Shared ASV Abundance: Among the gut microbiota containing shared ASVs with an abundance of ≥5%, the proportions of Streptococcus, Lactobacillus, and Klebsiella were notably elevated, while those of Faecalibacterium, Pseudomonas, Mucispirillum, and Parabacteroides were diminished.
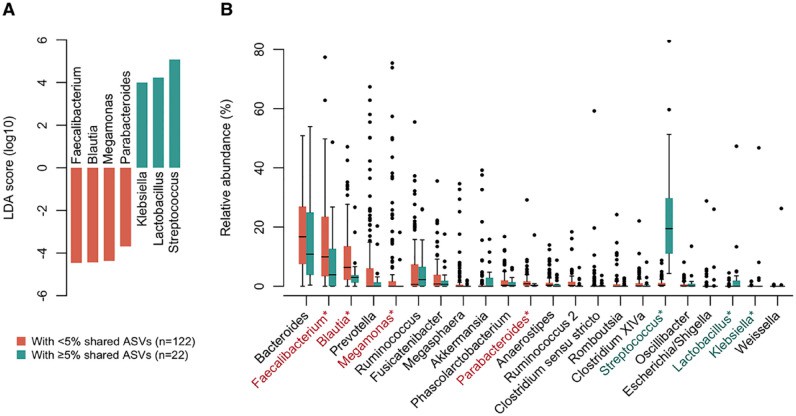 Figure 5. Gut microbiota composition associated with enrichment of oral bacteria.
Figure 5. Gut microbiota composition associated with enrichment of oral bacteria.
Significance of Findings: This high-resolution study provides evidence for the gut translocation of oral bacteria in community-dwelling adults. A significantly higher occupancy rate of shared ASVs was identified in elderly participants, indicating that the enrichment of oral bacteria in the gut is an age-related alteration.
The application of full-length microbial diversity sequencing in medical cohort studies facilitates enhanced resolution in species discovery. By identifying species-level microorganisms enriched in diseased groups and corroborating findings through experimental cultivation, researchers can more accurately pinpoint microbes associated with various diseases. This approach may further elucidate the interactive mechanisms between specific microbes and their host, thereby advancing our understanding of microbiota-related health implications.
References:


