The design and implementation of experiments are pivotal components of scientific research, particularly in the realm of gene mapping and genetic studies. A well-structured experimental design not only ensures the reliability and validity of data but also lays a foundation for subsequent analysis and interpretation. In gene mapping studies, the design of experiments significantly affects both the efficiency of the research process and the accuracy of its outcomes. Taking Bulk Segregant Analysis (BSA) as an example, an appropriately crafted experimental design can precisely pinpoint genes or genomic regions associated with specific traits.
BSA is a technique employed to swiftly identify markers linked to specific genes or genomic regions. The core principle of BSA involves combining individuals exhibiting extreme phenotypes to isolate the genetic variations associated with the traits of interest. The key components of experimental design in BSA include the selection of appropriate samples, the choice of suitable genetic markers, and the use of effective data processing methods. These elements work synergistically to ensure the reliability and precision of the experimental results.
This article aims to provide researchers with guidance on designing and implementing BSA experiments. By outlining the critical elements of BSA experimental design, this article assists readers in efficiently selecting samples, choosing markers, and employing data processing methods. Additionally, it introduces strategies to ensure the reliability of experimental outcomes through methodical experimental steps and validation processes. These insights provide robust support for the rapid and accurate identification of genes related to specific traits.
Key Steps in BSA Experimental Design
When designing a BSA experiment, critical steps include defining the research objectives and traits of interest, selecting appropriate parent lines and hybrid combinations, and proceeding through subsequent stages as detailed below:
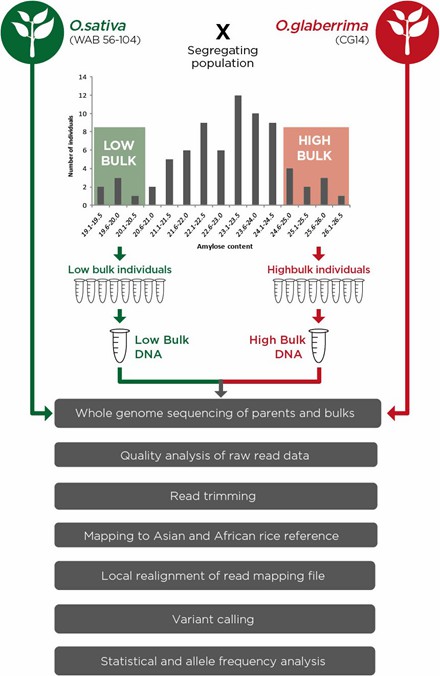 Schematic diagram of bulk segregant analysis experimental set-up and data analysis pipeline. (Wambugu, Peterson, et al. 2018)
Schematic diagram of bulk segregant analysis experimental set-up and data analysis pipeline. (Wambugu, Peterson, et al. 2018)
1. Define Research Objectives and Traits:
- The primary aim is usually to identify genes or markers associated with specific traits. For example, in the study of the root-knot nematode resistance gene Me3 in pepper, the goal is to pinpoint genes linked to nematode resistance.
- The choice of traits should be grounded in their significance for breeding or production. For instance, the research on the rice greening gene grc2 focuses on identifying genes influencing chlorophyll accumulation in rice leaves.
2. Select Appropriate Parental Lines and Hybrid Combinations:
- Choose parent lines with extreme phenotypes to ensure that the F2 population segregates for genetic variations linked to the target trait.
- Hybrid combinations should be chosen based on the genetic diversity between parents to enhance genetic variation within the F2 population.
3. Construct Bulk Segregant Populations:
- From the F2 population, select individuals exhibiting extreme phenotypes to form two bulk groups: one containing individuals with the target trait (e.g., resistant), and the other containing those without it (e.g., susceptible).
4. Conduct Molecular Marker Analysis:
- Employ molecular marker technologies such as SSR and SNP for genotyping the bulk populations to identify genetic markers associated with the target trait. In studies on genes affecting soybean seed coat color, SSR and SNP techniques were utilized for genotyping.
5. Perform Data Analysis:
- Through statistical analysis, compare the genotypic frequencies between the two bulk populations to identify genetic markers related to the target trait. For example, in the rice greening gene grc2 study, genotypic frequencies between extreme bulks were calculated to select relevant markers.
6. Verification and Further Analysis:
- Validate and extend the analysis of initially identified markers to confirm their association with the target trait. For instance, in wheat powdery mildew resistance gene research, chromosome positioning combined with BSA and microarray technology was used to conclusively map the resistance gene.
The success of BSA experiment design hinges on clearly defining research objectives and traits, selecting suitable parental lines and hybrid combinations, constructing bulk segregant populations, and identifying genetic markers related to the target trait through molecular marker analysis and statistical verification. These steps collectively form the core of BSA experiment design.
Services you may interested in
Want to know more about the details of BSA? Check out these articles:
Sample Selection and Processing in BSA
The selection and handling of samples are pivotal in research and must adhere to rigorous scientific methodologies. By clearly defining the target population, choosing an appropriate sampling frame and method, determining a reasonable sample size, and systematically collecting and preserving samples, researchers can ensure the representativeness and accuracy of their study outcomes.
Sample Selection
1. Define the Target Population:
- Initially, it is essential to define the target population, which encompasses the entire set of subjects or entities of interest in the study. This population could include specific individuals, items, or observation units that share common characteristics.
- For instance, in life sciences research, the target population might consist of specific cell types, tissues, or biological specimens.
2. Select a Sampling Frame:
- The sampling frame is a subset of the target population, typically represented as an accessible list or data source that reflects the entire population.
- In life sciences research, this could involve a registry of samples from specific cell types.
3. Choose the Sample:
- The sampling method depends on the research objectives, characteristics of the target population, and available resources. Common sampling techniques include simple random sampling, stratified sampling, systematic sampling, and convenience sampling.
- Stratified sampling is particularly useful for heterogeneous populations. It involves dividing the population into several mutually exclusive subgroups (strata) and randomly selecting samples from each subgroup.
- For example, in life sciences research, samples might be drawn from different cell types using stratified sampling to ensure representativeness.
4. Determine Sample Size:
- The sample size should be sufficiently large to facilitate robust data analysis, while remaining manageable.
- Determining sample size requires consideration of the study's aims, the characteristics of the target population, and the anticipated statistical power.
5. Implement the Sampling Plan:
- Once the sampling frame and method have been established, a detailed sampling plan should be devised. This plan should outline the selection of samples, the data collection process, and the procedures for recording and preserving samples.
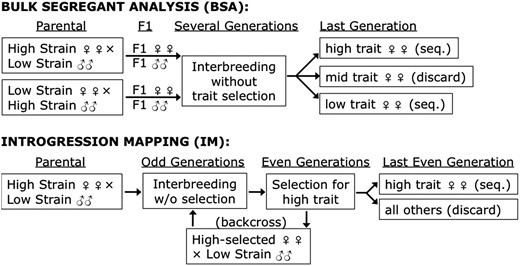 The investigated experimental designs for BSA and IM are illustrated. (Pool, John E., et al., 2016)
The investigated experimental designs for BSA and IM are illustrated. (Pool, John E., et al., 2016)
Sample Handling
1. Collect Samples:
- Methods for sample collection vary according to the type of research. In life sciences, samples may encompass serum, plasma, cell supernatants, cell lysates, urine, and tissue homogenates.
- Collection procedures must adhere to standardized protocols to ensure the quality and representativeness of the samples. For example, serum samples should be left at room temperature for 30–60 minutes (or longer) and then centrifuged at 3000 rpm for 30 minutes at 4°C.
2. Preserve Samples:
- Preservation methods depend on the sample type. Serum samples are typically stored at -20°C or -80°C.
- Other sample types, such as cell and tissue samples, require suitable preservation conditions to prevent degradation or contamination.
3. Data Collection and Recording:
- Concurrently with sample collection, detailed records of pertinent sample information should be maintained, including the source, collection time, collection method, and storage conditions.
- This information is crucial for subsequent data analysis and interpretation of results.
DNA Extraction and Sequencing for BSA
The selection of appropriate DNA extraction methods and sequencing technologies is vital for the success of BSA experiments. DNA extraction should be tailored to the sample type, with stringent quality control measures ensuring high purity and integrity of the DNA. The choice of sequencing technology should consider the sample type, target genome size, and budget, while leveraging efficient bioinformatics tools for data analysis. Adhering to these best practices can significantly enhance the success rate and accuracy of BSA experiments.
Best Practices in DNA Extraction
The quality of DNA extraction directly impacts the accuracy and reliability of subsequent sequencing results. Key considerations include:
- Sample Type and Processing: Select an extraction method appropriate for the sample type. For plant samples, the CTAB method is frequently employed. For microbial samples, using high concentrations of sodium dodecyl sulfate (SDS) for bead beating, followed by gentle DNA extraction and precipitation, is effective in preserving long DNA strands.
- Choice of Extraction Method: Different extraction methods can affect DNA quality and purity. For large-scale sample DNA extraction, the Midi-prep method is suitable. Additionally, selecting bioinformatics pipelines that align with the sample type and target genome is crucial. Algorithms and databases should be selected based on these criteria.
- Quality Control: Evaluate the concentration, purity, and integrity of extracted DNA using UV spectrometry and gel electrophoresis. For example, the Quant-iT PicoGreen dsDNA Assay Kit precisely measures DNA concentration, ensuring quality control.
Selecting Sequencing Technology
Next-Generation Sequencing (NGS) is commonly used in BSA experiments due to its ability to rapidly and efficiently analyze extensive genomic data. Consider the following when selecting sequencing technology:
- Choice of Sequencing Technology: NGS platforms, such as Illumina and Ion Torrent, vary in suitability depending on sample type, target genome size, and budget. The Illumina platform is widely suitable for sequencing most plant and animal samples.
- Sequencing Depth: Appropriate sequencing depth, determined by target genome size and variation type, ensures sufficient coverage. In rice research, for instance, high-throughput sequencing coupled with BSA can swiftly localize genes affecting specific traits.
- Data Analysis Tools: Selecting suitable bioinformatics tools for data analysis is critical. QTLseqr, an R package, facilitates the rapid identification of genomic regions associated with BSA.
Application Case Studies
- Plant Research: In rice, the combination of BSA and NGS successfully pinpointed genes influencing grain number. Similarly, in rapeseed, BSR-seq technology identified genes associated with yellow seed traits.
- Microbial Research: In yeast studies, BSA combined with NGS revealed genes linked to drug resistance.
- Crop Breeding: In soybean research, the integration of BSA with NGS accelerated the simultaneous identification of genes related to seed coat color.
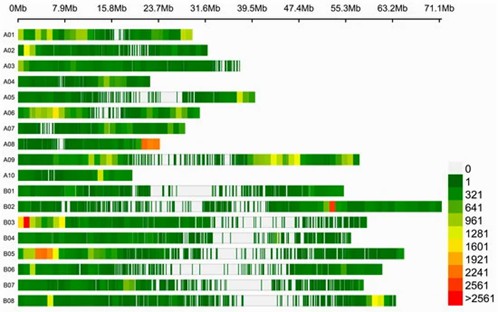 Distribution of SNPs/INDELs on 18 chromosomes. (Wang, Yang, et al., 2024)
Distribution of SNPs/INDELs on 18 chromosomes. (Wang, Yang, et al., 2024)
By meticulously selecting and applying DNA extraction methods and sequencing technologies, researchers can achieve precise and reliable results in BSA experiments, advancing our understanding of genetic traits across various organisms.
Want to know more about the details of BSA? Check out these articles:
Data Analysis and Interpretation in BSA
High-throughput sequencing technologies, coupled with advanced bioinformatics tools, enable a thorough analysis process from sequencing data to gene mapping. When interpreting BSA experiment results, it is imperative to consider statistical significance, candidate gene selection, functional validation, and the integration of multiple rounds of BSA results to ensure the accuracy and reliability of the final conclusions.
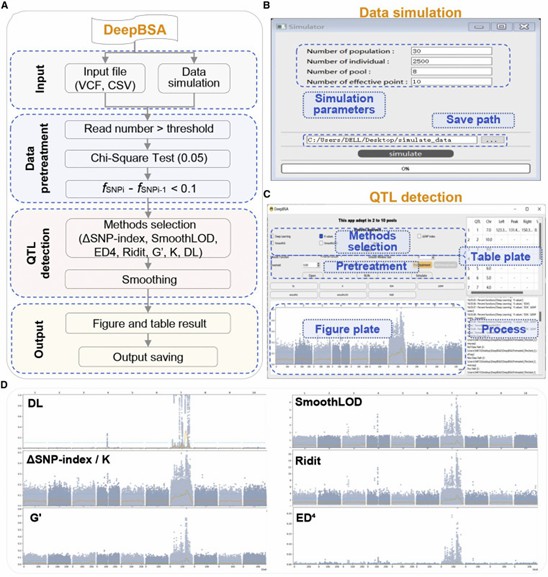 Graphical user interface of DeepBSA software. (Li, Zhao, et al. 2022)
Graphical user interface of DeepBSA software. (Li, Zhao, et al. 2022)
Workflow for Data Analysis:
1. Sample Preparation:
- Select two bulked populations exhibiting significant phenotypic differences: one comprising individuals with the target trait (e.g., disease resistance) and the other containing individuals without the trait (e.g., susceptibility).
- Perform sequencing on these bulked populations using high-throughput sequencing technologies, such as Next-Generation Sequencing (NGS).
2. Data Preprocessing:
- Conduct quality control on sequencing data, including the removal of low-quality reads and trimming of adapter sequences.
- Align sequencing reads to the reference genome using alignment tools like BWA.
3. Variant Detection:
- Identify variants (e.g., SNPs, InDels) and annotate them to determine their presence in target genes or genomic regions.
- Use statistical methods, such as the G statistic, to evaluate the distribution differences of variants between the two bulked populations.
4. QTL Mapping:
- Employ QTL analysis software (e.g., QTLseqr, PyBSA) to map the identified variants.
- Construct local genetic maps to pinpoint candidate genes associated with the target traits.
5. Result Validation:
- Further validate candidate genes through gene function annotation, expression analysis, and other methods.
- Confirm the association of candidate genes with the trait using marker-assisted selection techniques, such as SSR markers.
Interpretation of BSA Results:
1. Statistical Significance:
- Assess whether the detected variants exhibit statistical significance, indicating a significant distribution difference between the two bulked populations.
- Apply a smoothed version of the G statistic to account for sampling and sequencing-induced variations.
2. Candidate Gene Selection:
- Select candidate genes based on QTL mapping results, focusing on those tightly linked to the target trait.
- Evaluate the recurrence frequency of candidate genes across different experiments to enhance result reliability.
3. Functional Validation:
- Validate the association of candidate genes with the target trait through functional annotation and expression analysis.
- Further corroborate the linkage between candidate genes and the trait using marker-assisted selection techniques like SSR markers.
4. Multiple Rounds of BSA:
- If initial results are inconclusive, conduct additional rounds of BSA to refine the candidate region.
- Reconstruct bulked populations for each round and repeat the analysis workflow described above.
5. Integration of Results:
- Integrate results from various experiments and methods to build a comprehensive analytical framework.
- Consider discrepancies across different experimental conditions to ensure the robustness of the final conclusions.
Common Issues and Solutions in BSA Experiments
Common issues in Bulk Segregant Analysis experiments include sample contamination and data errors, as well as challenges in optimizing experimental design to improve success rates. These challenges can be effectively mitigated through standardized procedures, the use of sterile techniques, routine equipment calibration, careful determination of sample size, control of confounding variables, and appropriate data analysis methods, ultimately enhancing experimental outcomes and reliability.
1. Managing Sample Contamination and Data Errors
Sample Contamination
Sample contamination is a frequent issue in BSA experiments that can lead to biased results. The following strategies can help prevent and address sample contamination:
- Standardized Procedures: Ensure all experimental steps adhere to standardized operating procedures, covering sample collection, processing, and storage, to minimize human error and contamination risks.
- Sterile Techniques: Utilize sterile techniques and equipment throughout sample handling to prevent cross-contamination.
- Regular Calibration and Maintenance: Regularly calibrate and maintain laboratory equipment, such as centrifuges and pipettes, to ensure reliable performance.
- Sample Preservation and Transport: Promptly place collected samples under controlled conditions and transport them to the laboratory for analysis as soon as possible to prevent degradation.
Data Errors
Data errors can arise from various sources, including instrument malfunction, data processing mistakes, or transcription errors. Strategies to address data errors include:
- Outlier Detection: Utilize methods such as box plots, Grubbs' test, or PCA to identify outliers, and decide whether to remove or adjust them based on context.
- Data Reproducibility: Conduct multiple replicate experiments to verify consistency. This approach enhances data reliability and aids in identifying and rectifying potential errors.
- Data Analysis Tools: Master and employ suitable image processing and statistical tools to ensure accurate data interpretation and analysis.
2. Optimizing Experimental Design for Increased Success
Determining Sample Size
The size of the sample directly impacts the statistical significance of experimental results. Methods to optimize sample size include:
- Random Sampling: Select samples through random sampling to reduce bias and enhance representativeness.
- Increasing Sample Size: Where feasible, increase sample size to reduce sampling error and improve result reliability.
- Control of Confounding Variables: During experiment design, identify and control confounding variables (e.g., age, gender, health status) to ensure the accuracy of results.
Standardizing Experimental Design
A well-structured experimental design is crucial for ensuring success. Techniques to optimize experimental design include:
- Clear Experimental Objectives: Clearly define the questions or hypotheses the experiment aims to address before commencing.
- Selection of Appropriate Variables: Determine independent (manipulated) and dependent (responsive) variables, ensuring their selection is logical.
- Multiple Independent Replicates: Conduct multiple independent replicates of the experiment to assess result consistency and reliability.
Data Analysis Methods
Employing appropriate data analysis methods enhances the interpretive power of experimental results. Recommended data analysis approaches include:
- Descriptive and Inferential Statistics: Use descriptive statistics (e.g., mean, standard deviation) alongside inferential statistics (e.g., t-tests, ANOVA) for data analysis.
- Advanced Data Analysis Techniques: Utilize advanced methods such as regression analysis, and cluster analysis as needed to extract more information and support conclusions.
Conclusion
In designing and implementing Bulk Segregant Analysis experiments, success is contingent upon a comprehensive consideration of multiple factors. The selection of appropriate segregant populations is paramount, necessitating that samples exhibit significant phenotypic differences. High-throughput sequencing techniques, such as BSA-Seq and BSA-RNA-Seq, can substantially enhance the resolution and accuracy of these experiments. Statistical frameworks and algorithms, such as QTLseqr, provide robust support for data analysis, ensuring precise localization of quantitative trait loci (QTL).
Another critical aspect is the selection and validation of suitable molecular markers, such as single nucleotide polymorphisms (SNPs), to ensure the reliability of experimental results. Additionally, optimizing experimental design-particularly in terms of sample number, sequencing depth, and bulk size-can improve experimental outcomes. The integration of multi-omics data provides BSA experiments with more comprehensive insights into gene regulation, thereby deepening the scope of research.
Ultimately, results from BSA experiments require further validation and functional studies to confirm findings. By integrating other genetic analysis methods, researchers can enhance the reliability of the results. These carefully designed and implemented steps and methods offer crucial support for the rapid localization of genes influencing specific traits, thus advancing genetic improvement in plants and animals.
References:
- Bouso, Jennifer M., and Paul J. Planet. "DNA Extraction Method Optimized for Nontuberculous Mycobacteria Long-Read Whole Genome Sequencing." bioRxiv (2018): 470245. https://doi.org/10.1101/470245
- Gao, Yongbin, et al. "Conjunctive analyses of bulk segregant analysis sequencing and bulk segregant RNA sequencing to identify candidate genes controlling spikelet sterility of foxtail millet." Frontiers in Plant Science 13 (2022): 842336. https://doi.org/10.3389/fpls.2022.842336
- Wenger, Jared W., Katja Schwartz, and Gavin Sherlock. "Bulk segregant analysis by high-throughput sequencing reveals a novel xylose utilization gene from Saccharomyces cerevisiae." PLoS genetics 6.5 (2010): e1000942. https://doi.org/10.1371/journal.pgen.1000942
- Pool, John E. "Genetic mapping by bulk segregant analysis in Drosophila: experimental design and simulation-based inference." Genetics 204.3 (2016): 1295-1306. https://doi.org/10.1534/genetics.116.192484
- Kayam, Galya, et al. "Fine-mapping the branching habit trait in cultivated peanut by combining bulked segregant analysis and high-throughput sequencing." Frontiers in plant science 8 (2017): 467. https://doi.org/10.3389/fpls.2017.00467
- Wang, Yang, et al. "Identification of Yellow Seed Color Genes Using Bulked Segregant RNA Sequencing in Brassica juncea L." International Journal of Molecular Sciences 25.3 (2024): 1573. https://doi.org/10.3390/ijms25031573
- Wambugu, Peterson, et al. "Sequencing of bulks of segregants allows dissection of genetic control of amylose content in rice." Plant Biotechnology Journal 16.1 (2018): 100-110. https://doi.org/10.1111/pbi.12752
- Li, Zhao, et al. "DeepBSA: A deep-learning algorithm improves bulked segregant analysis for dissecting complex traits." Molecular Plant 15.9 (2022): 1418-1427. DOI: 10.1016/j.molp.2022.08.004

 Sample Submission Guidelines
Sample Submission Guidelines


