De novo assembly, in contrast to reference-guided assembly that relies on aligning sequencing reads back to an existing genome, is more independent and therefore not limited by reference biases. It is especially important for non-model organisms, large and complex genomes, or detecting novel sequences or structural variations. In spite of its transformative potential, de novo genome assembly represents a technically complex challenge, and it demands high sequencing depth alongside significant computational resources and advanced algorithms to deliver quality results. This approach is widely applicable, with implications for areas such as biodiversity science, evolutionary biology, and precision medicine, establishing it as an essential tool of contemporary genomics. De novo genome assembly provides unique flexibility, allowing the study of organisms for which no genome information exists. This complex process involves sophisticated computation and extensive resources.
Service you may interested in
Resource
Key Concepts and Challenges
- Repetitive Sequences: Repeats, which make up a large fraction of many genomes, are among the hardest challenges in genome assembly. Chimeras are formed from multiple reads that span multiple homology regions which are often subjected to preprocessing such as trimming and then leading to ambiguities in read placement and ultimately leading to assembly gaps and errors. For example, transposable elements can cause fragmented assemblies in eukaryotic genomes. Technologies that are capable of long-read sequencing will be critical in tackling this challenge, as they can cross these areas. Computational tools recognizing and masking repetitive sequences can also enhance assembly accuracy. Tools such as RepeatMasker are commonly used to identify repetitive regions, and applications such as Tandem Repeat Finder assist in the annotation of such sequences.
- Heterozygosity and Polyploidy: Many species are highly heterozygous or polyploid, making it difficult to differentiate and construct the existing alleles. The complexity is especially pronounced in plants, where polyploidy is widespread. To resolve these genomic features, they involve sophisticated algorithms that can resolve alleles or homeologous sequences and do so without losing their uniqueness. Assemblers like HiCanu are designed to handle such complexities, using long-read data to disentangle haplotype information.
- Sequencing mistakes: Different sequencing technologies have different mistakes profiles long-read technologies like PacBio and Oxford Nanopore, for instance, tend to yield higher error rates than short-read platforms. These are errors can lead to false assembles or calls. Error correction tools for short-read assembly refinement or long-read datasets are vital for obtaining high-quality assemblies. In addition, hybrid assembly strategies utilize the advantages of both short and long reads to alleviate the above problems.
- Data and Algorithms: De novo genome assembly involves the handling of vast amounts of data, and the execution of very complex algorithms. To address these limitations, high-performance computing resources such as running parallelized workflows and even using cloud computing platforms are becoming more common. Memory and algorithm usage remains an active area of research. One approach is to minimize computation while maintaining assembly quality, and several assemblers exemplify this strategy.
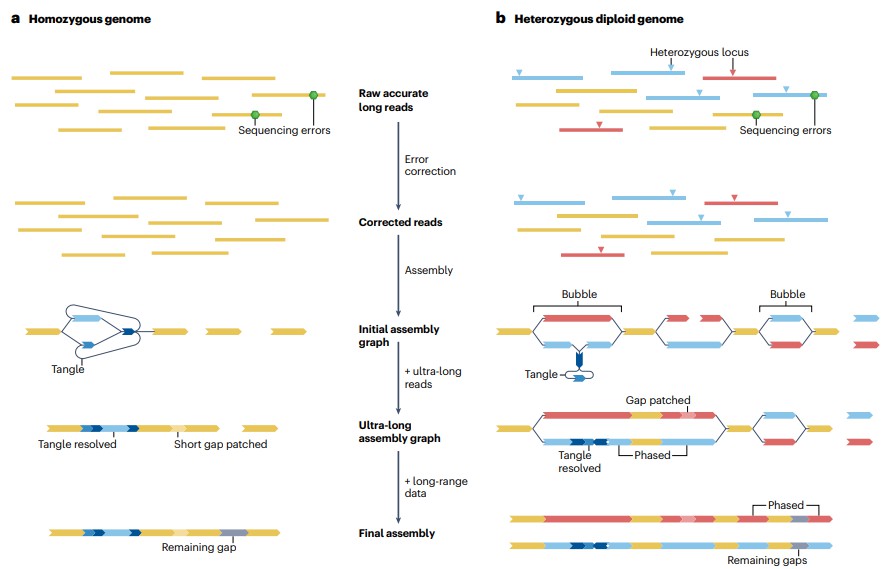 Strategy for near-telomere-to-telomere assembly (Li H, Durbin R., 2024).
Strategy for near-telomere-to-telomere assembly (Li H, Durbin R., 2024).
Technologies and Methods
Sequencing Platforms
Long-read platforms, e.g., PacBio and Oxford Nanopore, generate reads extending tens to hundreds of kilobases, allowing the resolution of repetitive elements and large structural variations. Such technologies are essential for complex or polyploid genomes. However, continuous advancements in chemistry and base-calling algorithms are shrinking the gap in accuracy despite a higher error rates. PacBio HiFi reads, for instance, now combine high read length and high accuracy and are a favorite for a lot of assemblies.
Assembly Algorithms
- Overlap-Layout-Consensus (OLC): Specifically designed for and works great on long-read data. It finds overlaps between reads, constructs a layout graph, and derives consensus sequences. Assembly of large genomes using Overlap-Layout-Consensus (OLC) was pioneered by tools such as Canu and FALCON. The OLC genotyping more accurately than GFA for the complex genomic regions.
- De Bruijn Graphs (DBG): DBG methods offer high computational efficiency and work best with datasets generated from short sequencing reads. DBG approaches tokenize reads into k-mers and build a graph in which paths are genomic sequences. However, repetitive regions can lead to complicated graph structures which require specialized algorithms to resolve. Common DBG-based assemblers for small to medium genomes include Velvet and SOAP denovo.
- Modern Hybrid Assemblers: By combining the benefits of both OLC and DBG, hybrid assemblers take assembly workflows to the next level utilizing both short- and long-read datasets. Such a strategy provides high contiguity, completeness, and accuracy despite difficult genomes. Hybrid assembly techniques, such as those employed by SPAdes and Flye, can prove powerful, especially in the case of genomes with high repeat content or complex structural variation.
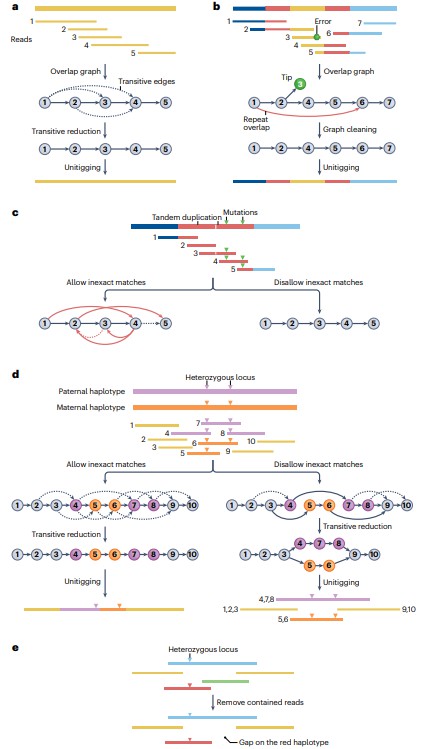 Assembly with overlap graphs (Li H, Durbin R. et al, 2024).
Assembly with overlap graphs (Li H, Durbin R. et al, 2024).
De Novo Genome Assembly Steps
The de novo genome assembly procedure consists of a series of interconnected stages, where each step is an important contributor towards the generation of a high-quality final assembly. Attention must be paid to each of these stages, from data preparation through validation, to result in credible and accurate outcomes.
Data Preparation
By far the most important is the quality of the data used for assembly — without high-quality data, any assembly project is doomed. Preprocessing steps include:
- Quality control, filtering and trimmig : Raw sequencing data is checked for quality using tools like FastQC and low quality reads, adapters and contaminants are identified. Trimmers such as Trimmomatic and Cutadapt trim unwanted parts to assure only high confidence reads go for the assembly. This step is critical in removing low-complexity regions prone to generating dust in the assembly.
Assembly Construction
Genome assembly is an iterative process:
- Contig Generation: The short reads are assembled into contigs, which are the longest contiguous sequences, or stretches, that can be produced without gaps. To achieve the highest possible accuracy and contiguity, you can use specialized tools such as Canu (long reads) and Velvet (short reads) that are used for this purpose.
- Scaffolding: Contigs are joined into scaffolds based on paired-end or long-read data. Scaffolding adds spatial information that orders and orients the contigs correctly. SSPACE and BESST are commonly employed to help improve structural accuracy of assembly.
- Gap Closing: Gaps in scaffolds are filled using additional sequencing data or computational algorithms such as GapCloser to improve continuity. Segmentation with Accurate gap fillings guarantee deeper completeness which contains less frequent sequence.
Assembly Validation
Assembly validation ensures completeness and correctness:
- Metrics: N50 to evaluate contiguity of assembly and BUSCO to assess completeness using conserved sets of single-copy orthologs. These metrics serve as quantitative indicators of assembly quality and help in refining the assembly process further.
- Validation Tools: QUAST produces detailed quality assessments, pinpointing misassemblies and opportunities for enhancement. Using the REAPR tool, structural inconsistencies are detected, requiring refinement to yield a more reliable assembly.
Future Directions and Applications
De novo genome assembly has a range of applications, from basic research to applied sciences, and even more future developments will impetus its potential.
Applications
- Non-Model Organisms: Supply genomic context for species without reference genomes via de novo assemblies. These tools are invaluable for the study of biodiversity, the discovery of new genes, and the investigation of evolutionary adaptations. For example, by reconstructing the genome of extremophilic organisms, we have uncovered pathways unique to these organisms that allow them to survive in extreme conditions.
- Conservation Biology: Genome assemblies guide conservation by elucidating genetic diversity, population structure, and inbreeding levels in endangered species. This information is essential for good management and breeding programs. And his study of species assemblages, such as the giant panda, has offered important perspectives regarding their evolutionary history and adaptive processes.
- Medicine: Novel virulence factors, drug-resistance mechanisms, and other evolutionary and epidemiological discoveries are enabled by the de novo assemblies of pathogens. The sequencing of SARS-CoV-2, for example, has been critical for developing vaccines and tracking outbreaks. De novo assemblies of individual genomes also benefit personalized medicine by revealing unique structural variations and mutations.
Future Directions
However, there are computer algorithms that can only work on long-read sequencing data and these datasets will be necessary because they will enable us to resolve complex regions in the genome, such as the centromeres and telomeres. These improvements will probably update the definition of assembly completeness.
Machine learning: The addition of machine-learning algorithms in assembly pipelines provides corrections for errors and also results in more repeat variations and structural variants, providing higher accuracy and efficiency. AI-derived tools are also hastening the creation of adaptive algorithms customized to particular genomic tasks. For a more in-depth understanding of machine-learning algorithms, refer to our article "Genome Indexing in Bioinformatics: Unpacking the Genome".
- Genomics at Single-Cell Resolution: Genotyping to Haplotype-resolved assemblies: Single-cell sequencing has the potential to provide the necessary resolution to identify haplotype-resolved assemblies, and can lead the way to understanding genetic heterogeneity and evolutionary processes in populations. This is especially relevant in cancer research given that intra-tumor heterogeneity is known to be of great importance in disease evolution.
- Standardization and Sharing: Introduction of standardized workflows and open-access databases will enhance reproducibility and promote collaboration, maximizing the contributions of assembled genomes. The Earth BioGenome Project and similar efforts hope to create a full, curated reference library for the genomes of all known eukaryotic species, containing in one place similar sequences needed for the study of this diversity and its relationship to any given habitat or niche.
Case Study: The Giant Panda Genome Assembly
Background
The giant panda, an iconic species, has been a focus of conservation efforts due to its endangered status and ecological significance. Understanding its genetic makeup is critical for designing effective conservation strategies, assessing genetic diversity, and studying its unique adaptations, such as a bamboo-dominated diet. However, the giant panda genome posed unique challenges due to its high repeat content and low genetic variability. Here is the case for the application of De novo genome assembly in giant panda genome assembly.
Methods
To achieve a high-quality assembly of the giant panda genome, researchers employed the following steps:
- Sequencing Technologies: A hybrid sequencing approach was used. Short-read sequencing with Illumina provided high-accuracy reads, while long-read sequencing from PacBio resolved repetitive regions and enabled the assembly of complex genomic structures.
- Assembly Algorithms: De novo assembly was performed using the SOAPdenovo assembler, optimized for large genomes with high repeat content. Additional scaffolding tools, including SSPACE, improved the contiguity and accuracy of the assembly. RepeatMasker was employed to annotate and mask repetitive elements, while Pilon polished the assembly to correct base-level errors.
- Validation and Annotation: Assembly quality was evaluated using metrics such as N50 and BUSCO scores. Gene prediction tools, including AUGUSTUS and MAKER, were applied to annotate coding sequences and regulatory elements. Comparative genomic analyses with related species further validated the assembly.
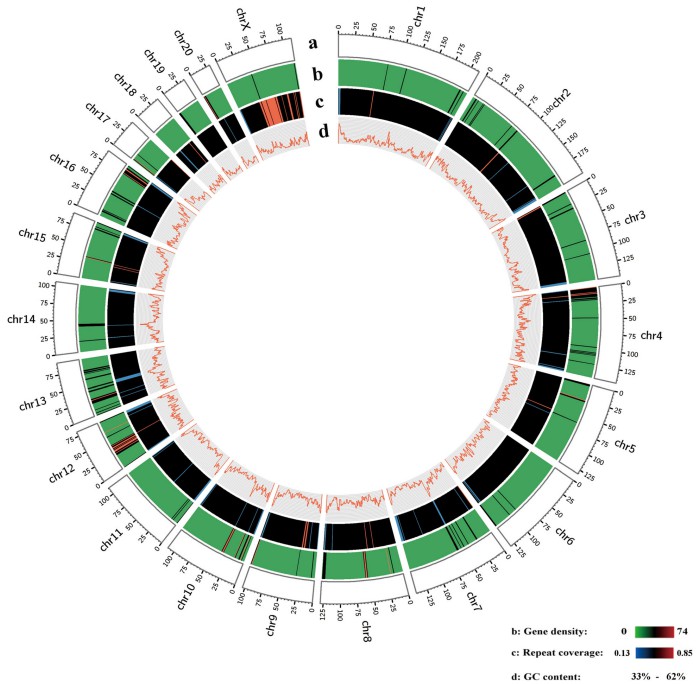 Characterization of the giant panda genome landscape (Fan, H. et al, 2019).
Characterization of the giant panda genome landscape (Fan, H. et al, 2019).
Results
The de novo assembly of the giant panda genome achieved a contig N50 of approximately 200 kb and scaffold N50 of over 1 Mb, representing a high level of continuity and completeness. Key findings included:
- Identification of genes linked to bamboo digestion, such as those involved in cellulose breakdown.
- Insights into the genetic basis of its low reproductive rate and immune system adaptations.
- High-resolution mapping of genetic diversity within wild and captive populations, informing conservation programs.
Conclusion
De novo genome assembly is a linchpin of modern genomics and continues to enable investigations of genetic architecture across a wide variety of organisms. This technology has transformed fields from evolutionary biology to precision medicine, and addressed challenges like repetitive sequences, heterozygosity, and computational load. De novo assembly continues to broaden its horizons with technological and computational improvements. However, as we shift towards genomics becoming more prevalent and widespread, the knowledge acquired from de novo genome assembly will be fundamental to solving many of the challenges facing the world today, and expanding our comprehension of life itself.
References:
-
Li, H., & Durbin, R. (2024). Genome assembly in the telomere-to-telomere era. Nature reviews. Genetics, 25(9), 658–670. https://doi.org/10.1038/s41576-024-00718-w
- Fan, H., Wu, Q., Wei, F., Yang, F., Ng, B. L., & Hu, Y. (2019). Chromosome-level genome assembly for giant panda provides novel insights into Carnivora chromosome evolution. Genome biology, 20(1), 267. https://doi.org/10.1186/s13059-019-1889-7

 Sample Submission Guidelines
Sample Submission Guidelines


