We explores genome-wide linkage analysis (GWLA), an essential tool for uncovering genetic factors associated with traits and diseases through family-based studies. GWLA focuses on co-segregation patterns within families, making it particularly effective for identifying rare or novel variants often overlooked by genome-wide association studies (GWAS).
We begin by explaining the principles and methodology of GWLA, highlighting its advantages over GWAS. The document then provides a step-by-step guide to conducting a GWLA study, from study design and genotyping to data analysis. Practical applications are discussed, including its role in disease gene discovery, functional genomics, and advancements in agricultural and animal genetics. A case study on hereditary breast cancer demonstrates GWLA's real-world impact, showcasing how the BRCA1 gene was identified.
Finally, we review future directions, emphasizing the integration of GWLA with modern genomic technologies like next-generation sequencing. This comprehensive summary underscores GWLA's ongoing relevance and its contributions to advancing genetics across diverse fields.
What is Genome-Wide Linkage Analysis
GWLA is a powerful genetic mapping approach to identify linked chromosomal regions with the manifestation of the trait or disease. Its foundation lies in genetic linkage theory, the idea that alleles that are physically close to one another on a chromosome are likely to be inherited together. This bias occurs because recombination events during meiosis are less effective in splitting closely linked alleles so that they co-segregate more frequently than alleles far apart.
The success of GWLA relies heavily on its ability to analyze recombination events, which generate distinct inheritance patterns in family pedigrees. Statistical models, such as logarithm of odds (LOD) scores, are then applied to assess the likelihood of linkage between markers and the trait under investigation. A high LOD score indicates a strong correlation, suggesting that the linked region is worth further exploration.
Unlike GWAS, which focus on unrelated individuals and common variants, GWLA delves into familial inheritance to detect rare or novel variants. This makes GWLA an invaluable tool for identifying genetic factors that might be missed by population-based studies, particularly in conditions with clear familial aggregation or when studying traits influenced by rare genetic variants.
Service you may intersted in
How to Do Genome-Wide Linkage Analysis
Genome-wide linkage analysis remains an indispensable tool in genetic research, offering unique insights into the genetic architecture of diseases and traits. Unlike GWAS, which is limited in its ability to detect rare variants, GWLA excels in identifying rare and novel loci that may underlie familial patterns of inheritance. By focusing on co-segregation within families, GWLA provides a powerful framework for exploring the genetic basis of complex traits, particularly those influenced by rare variants or strong genetic determinants.
The workflow for pedigree analysis in GWLA can be summarized as follows:
Study Design
- Recruit Families: Select families with multiple affected individuals to maximize the power of detecting linked regions. Both affected and unaffected members should be included.
- Phenotype Definition: Precisely define the trait or disease under investigation. Phenotypes should be measurable and consistent across individuals.
Genotyping
- Marker Selection: Use polymorphic markers, such as SNPs or microsatellites, distributed evenly across the genome.
- Genotyping Platforms: Employ high-throughput technologies to genotype markers in all family members.
Data Quality Control
- Ensure high call rates and remove markers with low minor allele frequency (MAF) or poor quality.
- Verify familial relationships through kinship analysis to detect and resolve pedigree errors.
Linkage Analysis
- Statistical Methods: Apply either parametric (model-based) or non-parametric (model-free) methods to assess linkage.
- Parametric Linkage Analysis: Requires a genetic model specifying mode of inheritance, penetrance, and allele frequencies.
- Non-parametric Linkage Analysis: Evaluates allele sharing among affected individuals without requiring a predefined genetic model.
- LOD Score Calculation: Compute the LOD score to assess the likelihood of linkage. A LOD score ≥3 is typically considered evidence of significant linkage.
Interpretation and Fine Mapping
- Identify chromosomal regions with significant LOD scores.
- Conduct fine mapping and functional studies to narrow down candidate genes.
- careful peak deconvolution and identification processes.
- NMR Spectroscopy: NMR spectroscopy is a non-destructive technique that provides structural information about metabolites. It is particularly useful for quantifying metabolites without extensive sample preparation. NMR data, however, can be challenging to interpret due to overlapping signals and the need for expertise in spectral analysis.
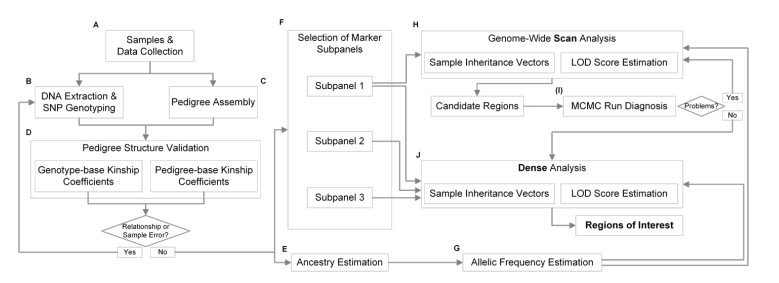 Schematic diagram of the pedigree analysis (GWLA) mapping workflow. (Magalhães Borges, V. et al., 2024)
Schematic diagram of the pedigree analysis (GWLA) mapping workflow. (Magalhães Borges, V. et al., 2024)
Genome-Wide Linkage Analysis vs. GWAS
While GWLA and GWAS are both genome-wide approaches, they have fundamental differences in methodology, scope, and applications:
| Aspect |
GWLA |
GWAS |
| Study Population |
Families (related individuals) |
Unrelated individuals |
| Trait Focus |
Mendelian traits or rare variants |
Complex traits with common variants |
| Markers Used |
Polymorphic markers (e.g., SNPs, microsatellites) |
SNP arrays or whole-genome sequencing |
| Inheritance Model |
Relies on recombination and co-segregation |
Relies on allele frequency differences |
| Statistical Approach |
LOD scores for linkage |
p-values for association |
| Key Output |
Linked chromosomal regions |
Associated SNPs |
| Power to Detect Rare Variants |
High |
Low |
Differences between GWLA and GWAS. (Yuan J et al., 2019)
GWLA is ideal for discovering novel loci in diseases with clear familial aggregation, while GWAS excels in identifying common risk alleles across diverse populations. In some studies, these methods are combined to provide complementary insights.
Applications of GWLA
GWLA has diverse applications in genetics and biomedical research. Here, we briefly provided some area that can apply GWLA:
Disease Gene Identification
Successfully used to identify genes for Mendelian disorders, such as cystic fibrosis (CFTR gene) and Huntington's disease (HTT gene).
Provides initial leads for studying complex diseases like schizophrenia, diabetes, and cardiovascular disorders.
GWLA has played a pivotal role in advancing precision medicine. By identifying genetic factors associated with rare disorders, it enables early diagnosis, targeted interventions, and tailored therapeutic strategies. For instance, discoveries made through GWLA have been instrumental in developing gene-based therapies, such as those targeting cystic fibrosis and sickle cell anemia.
Rare Variant Discovery
Efficient for uncovering rare or family-specific variants that may be missed in population-based approaches.
Helps link genetic loci to biological pathways and functions, facilitating a deeper understanding of gene-disease mechanisms.
Pharmacogenetics
Identifies genetic factors influencing drug response, aiding in the development of personalized medicine.
Animal and Plant Genetics
Widely applied in agricultural genetics to map traits like disease resistance, yield, and drought tolerance in crops and livestock.
Beyond human health, GWLA has significantly contributed to agricultural genetics. It has facilitated the identification of genes associated with traits like pest resistance, drought tolerance, and yield improvement in crops. Similarly, in livestock, GWLA has helped map genetic determinants of traits such as disease resistance and growth rates, supporting more efficient breeding practices.
Integration with Modern Technologies
The utility of GWLA has been further enhanced by advancements in sequencing technologies and bioinformatics. Next-generation sequencing (NGS) now allows researchers to fine-map linkage regions with unprecedented precision, enabling the identification of causal variants at the nucleotide level. Additionally, combining GWLA with other approaches, such as GWAS and transcriptomics, creates a synergistic platform for understanding the interplay between genetic, epigenetic, and environmental factors in shaping phenotypes.
Genome-wide linkage analysis remains a powerful tool in the geneticist's toolkit. By providing insights into the genetic basis of diseases, it complements modern approaches like GWAS and continues to drive advances in human health, agriculture, and beyond.
Case Study of GWLA
Here, we provide an example for the application of GWAL in identifying a Gene for Breast Cancer. Literature resources: Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. (Zhang, H. J et al., 2020)
Breast cancer is of the significant health concern worldwide. Early detection of genetic link in families of breast cancer is of great clinic significance. Researchers suspected a genetic link in families with multiple cases of early-onset breast cancer. Families with a strong history of breast cancer were recruited. Detailed pedigrees were constructed, and blood samples were collected for DNA analysis.
Genotyping: Microsatellite markers spanning the genome were genotyped in affected and unaffected family members. Parametric linkage analysis was performed assuming autosomal dominant inheritance. Significant LOD scores were observed on chromosome 17q. The linked region was narrowed down using additional markers. Sequencing identified mutations in the BRCA1 gene within the linked region. Functional studies confirmed its role in DNA repair.
This discovery revolutionized breast cancer research, leading to genetic testing and risk management strategies for carriers of BRCA1 mutations. It also highlighted the utility of GWLA in uncovering critical disease-associated genes.
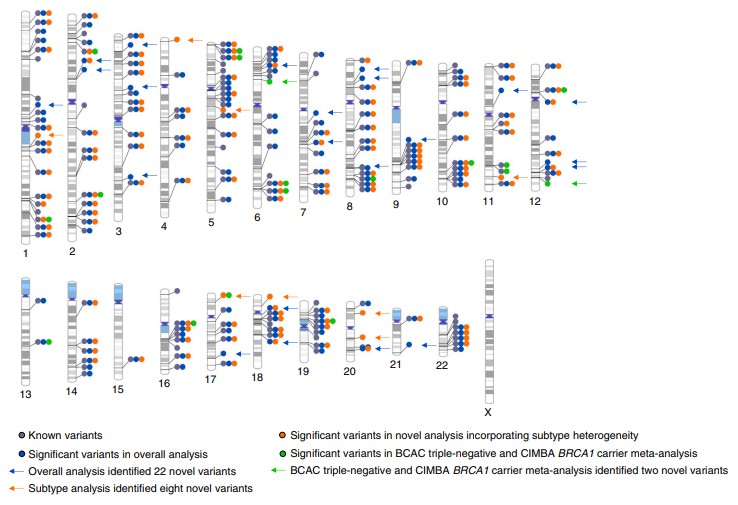 Diagram of all distinct genome-wide-significant variants. (Zhang, H. J et al., 2020)
Diagram of all distinct genome-wide-significant variants. (Zhang, H. J et al., 2020)
References:
-
Magalhães Borges, V., Horimoto, A. R. V. R., Wijsman, E. M. et al. (2024). Genomic Exploration of Essential Hypertension in African-Brazilian Quilombo Populations: A Comprehensive Approach with Pedigree Analysis and Family-Based Association Studies. medRxiv : the preprint server for health sciences, 2024.06.26.24309531. https://doi.org/10.1101/2024.06.26.24309531
- Yuan, J., Tickner, J., Mullin, B. H. et al. (2019). Advanced Genetic Approaches in Discovery and Characterization of Genes Involved With Osteoporosis in Mouse and Human. Frontiers in genetics, 10, 288. https://doi.org/10.3389/fgene.2019.00288
- Zhang, H., Ahearn, T. U., Lecarpentier, J. et al. (2020). Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nature genetics, 52(6), 572–581. https://doi.org/10.1038/s41588-020-0609-2

 Sample Submission Guidelines
Sample Submission Guidelines


