
 Sample Submission Guidelines
Sample Submission Guidelines
The Introduction of Pan Genome Sequencing
The concept of the pan-genome encompasses the entirety of genes present across all strains within a given species, thereby offering a comprehensive perspective on genetic diversity that transcends the limitations of individual genome sequences. It includes two primary components:
- Core Genome: This segment comprises genes ubiquitously present across all strains of the species. These core genes are typically associated with fundamental biological functions and primary phenotypic traits, thereby reflecting the evolutionary stability of the species. For instance, in Escherichia coli, core genes include those implicated in essential cellular processes such as DNA replication and transcription.
- Variable (Accessory/Dispensable) Genome: In contrast, this segment encompasses genes that are found in only a subset of strains or individuals within the species. These accessory genes often encode for specific adaptations or unique traits, including factors such as antibiotic resistance or virulence. For example, in Streptococcus pneumoniae, accessory genes contribute to variations in pathogenicity and resistance profiles among different strains.
Through the comprehensive study of the pan-genome, researchers can gain insights into the full spectrum of genetic variability within a species, facilitating a deeper understanding of its evolutionary dynamics, functional adaptations, and potential response to environmental pressures.
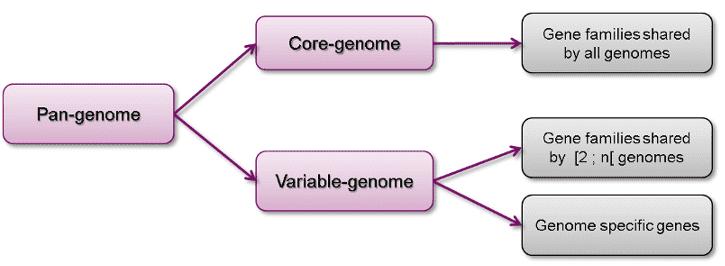 Figure 1. Composition of a pan genome
Figure 1. Composition of a pan genome
The pursuit of pan-genome sequencing involves leveraging high-throughput sequencing technologies alongside sophisticated bioinformatics tools. This approach entails the meticulous construction of sequencing libraries followed by comprehensive sequencing of individuals, subspecies, or lineages within a species. Subsequent assembly of these sequences leads to the development of a pan-genome map. This map serves to enrich the genetic repository of the species and enables the investigation of crucial biological questions, contributing significantly to our understanding of genetic diversity and evolutionary processes.
Why is Pan-Genome Research Essential
In the extensive course of evolution, influenced by geographical and environmental factors, individual organisms develop highly unique genetic traits. A single reference genome is insufficient to encompass the complete genetic information of an entire species. In other words, relying solely on a single reference genome for studies on genetic domestication and variation can result in the loss of significant genomic content, as many unique sequences are not represented in the reference genome.
Moreover, the decreasing cost of genome sequencing has made pan-genome research increasingly viable in recent years. This reduction in cost facilitates the exploration of genetic diversity at a depth and scale that was previously unattainable. Consequently, the study of pan-genomes has become a burgeoning field, offering profound insights into the complexity of genomic information and the adaptive potential of species.
Difference Between the Pan-Genome and the Whole Genome
The concept of the whole genome pertains to the complete set of genetic material found within a single individual or strain, typically leveraged as a reference in genomic research. In contrast, the pan-genome amalgamates genetic information from multiple individuals or strains, offering a more holistic and comprehensive genetic landscape of the species.
Key Differences
- Scope: The whole genome delineates the genetic architecture of a single strain. By contrast, the pan-genome encapsulates all genetic variants present across multiple strains, thereby capturing a broader and more inclusive spectrum of genetic diversity.
- Resolution: Pan-genome analysis elucidates genetic variations that may remain undetected when relying solely on a single reference genome. This encompasses unique accessory genes that drive strain-specific traits and adaptations.
For instance, examination of the pan-genome of Mycobacterium tuberculosis reveals considerable genetic variability among different strains, a factor of paramount importance for understanding mechanisms of drug resistance and for the development of effective therapeutic interventions.
Advantages of Pan Genome
- Enrich genome information of the species through sequencing the subspecies and individuals
- Quickly find genes or structural variations of genes related to important traits based on study of variable genome
- Study the differences within species from the perspective of unique gene sequences
- Small population, cost and time efficient
Applications of Pan Genome
- Species Evolution and Phylogenetics: Assists in tracing evolutionary relationships and genetic diversity among species.
- Trait Discovery and Breeding: Identifies genes associated with desirable traits for improved crop breeding.
- Pathogen Research: Reveals genetic variations in pathogens related to virulence and resistance, informing control measures.
- Ecological and Environmental Studies: Provides insights into species' adaptations to different environments and ecological niches.
Pan Genome Workflow

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
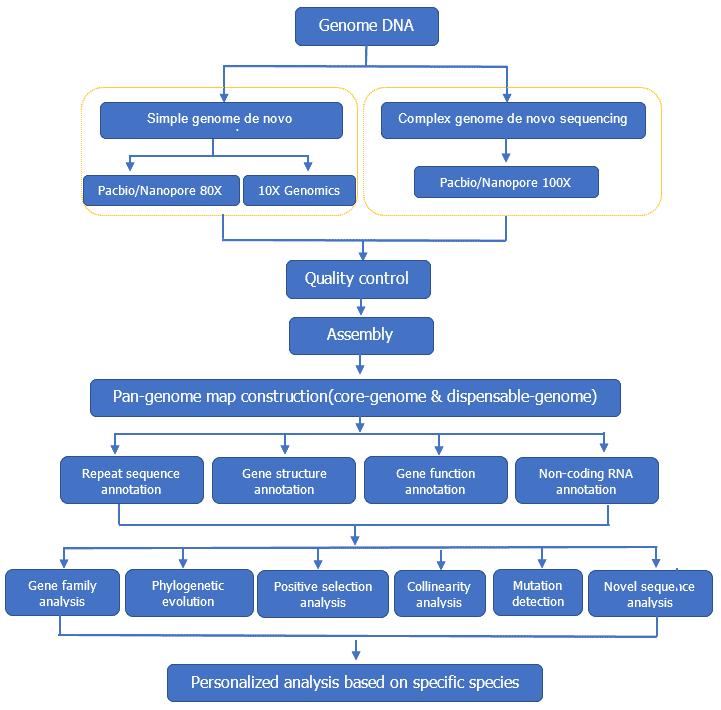
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Pan Genome for your writing (customization)
Partial results are shown below:
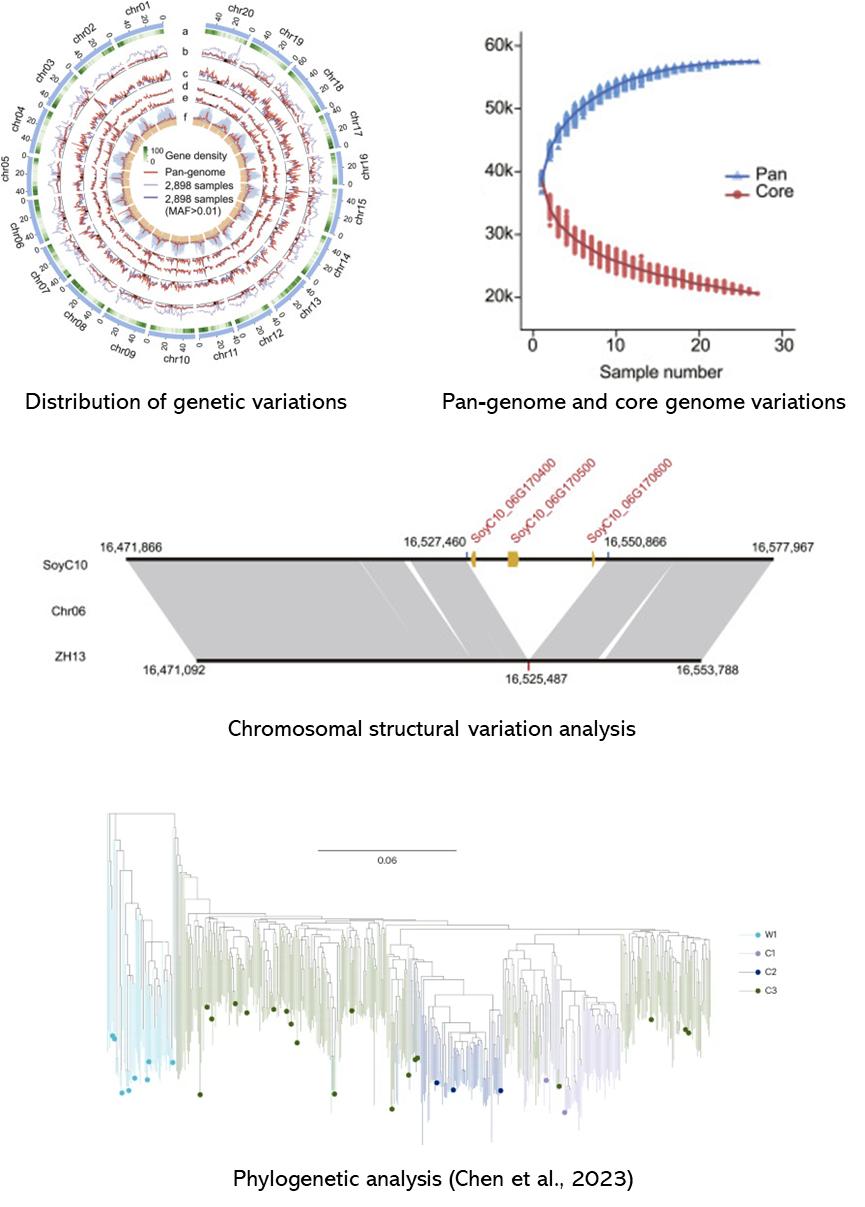
References
- Liu Y, Du H, Li P, et al. Pan-genome of wild and cultivated soybeans. Cell. 2020, 182(1):162-76.
- Chen J, Liu Y, Liu M, et. Pangenome analysis reveals genomic variations associated with domestication traits in broomcorn millet. Nature Genetics. 2023, 55(12):2243-54.
1. How many samples are required for pan-genome sequencing?
A minimum of two individuals or subspecies is essential to commence a pan-genome analysis. However, to achieve a more exhaustive understanding of genetic diversity within a species, a larger number of samples should be considered. The inclusion of additional samples significantly enhances the resolution of genetic variation and leads to a more comprehensive pan-genome map.
2. Is a reference genome required for pan-genome analysis?
The utilization of a reference genome in pan-genome analysis is not strictly obligatory but can be beneficial. A reference genome serves as an initial framework for gene annotation and comparative analysis. Nonetheless, the primary objective of pan-genome studies is to uncover genetic variations that transcend the scope of a single reference genome. Therefore, pan-genome analysis is designed to identify and incorporate genetic diversity beyond that represented by any single reference, ensuring a holistic capture of the species' genomic landscape.
3. What types of analyses are performed in standard pan-genome services?
Pan-Genome Assembly: Includes GC content analysis, sequencing depth analysis, and construction of super-scaffolds using reference genomes.
Pan-Genome Structural Annotation: Involves predicting core and accessory genomes, annotating repeat sequences, and identifying non-coding RNAs.
Pangenome analyses reveal impact of transposable elements and ploidy on the evolution of potato species
Journal: Proceedings of the National Academy of Sciences
Impact factor: 12.779
Published: July 24, 2023
Background
Potato (Solanum tuberosum) is a key global crop with rich genetic diversity. Originating in the Andean highlands, it spans multiple ploidy levels. This study presents the most comprehensive potato pan-genome, combining sequences from 296 accessions and identifying 132,355 pangenes. The analysis highlights genetic diversity, adaptation, and evolutionary relationships within the Solanum section Petota.
Materials & Methods
Sample Preparation
- Potato
- Genome sequences of potato accessions
Method
- Sequencing data download
- Construction of the pangenome
- De novo assembly
- Alignment
- Annotation of the pangenome
- PAV analysis
- Gene frequency differences analysis
Results
1. The Pangenome of Solanum Section Petota
The pangenome comprises 296 potato samples with diverse origins and ploidy levels, including 154 Gbp of new and public assemblies, resulting in 514,888 contigs and 132,355 gene models.
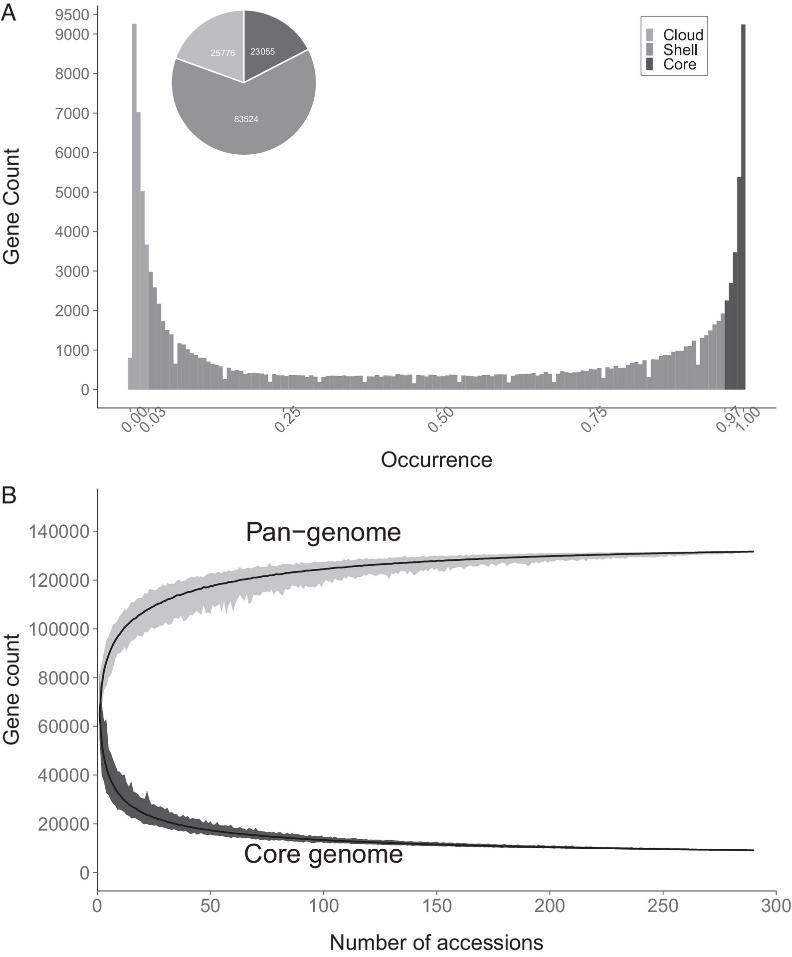 Fig 1. The Solanum section Petota pangenome.
Fig 1. The Solanum section Petota pangenome.
2. PAV Variation in Protein-Coding Genes
Variation in protein-coding genes across samples is influenced by ploidy and domestication, affecting gene numbers and selection.
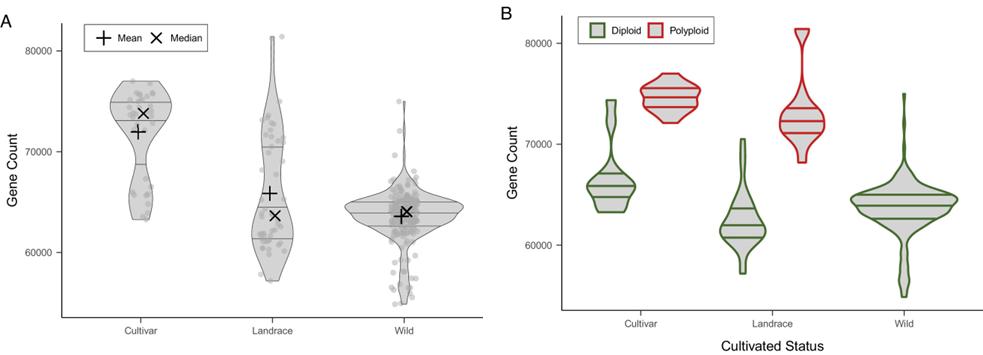 Fig 2. Gene content of the accessions included in the Solanum section Petota pangenome based on PAV.
Fig 2. Gene content of the accessions included in the Solanum section Petota pangenome based on PAV.
3. Clustering the Accessions Based on Gene Content
Phylogenetic analysis and PCA identify distinct clades and subgroups, reflecting gene content variations.
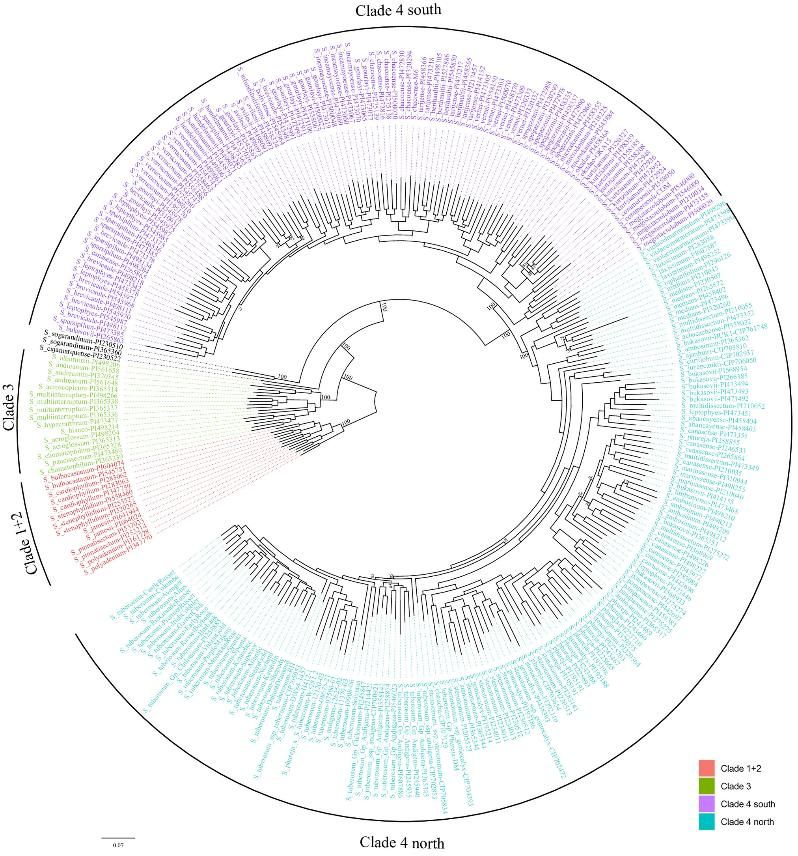 Fig 3. A maximum likelihood phylogenetic tree constructed using PAV data for the Solanum section Petota pangenome.
Fig 3. A maximum likelihood phylogenetic tree constructed using PAV data for the Solanum section Petota pangenome.
4. Variation in Gene Content Distinguishes Clades and Subgroups
Gene content differences are notable between clades, highlighting evolutionary and adaptation traits.
5. TE Content in the Pangenome
The pangenome contains 75.5% transposable elements, with significant clade-specific variation and differences in TE content.
Conclusion
Future potato crop survival amid climate change relies on conserving biodiversity and integrating it into new cultivars. A pangenome of 296 accessions from 60 Solanum section Petota species reveals that PAV, especially TE variation, plays a role in speciation and shows increased TE content in in vitro propagated materials, similar to natural stress-induced TE activity.
Reference
- Bozan I, Achakkagari SR, Anglin NL, et. Pangenome analyses reveal impact of transposable elements and ploidy on the evolution of potato species. Proceedings of the National Academy of Sciences. 2023, 120(31):e2211117120.
Here are some publications that have been successfully published using our services or other related services:
Collection of genetic data in ethnic-based studies across Aymaras, Quechuas and Mestizos: the challenges of the Genetics of Alzheimer's in Peruvian Population (GAPP) study
Journal: Alzheimer's & Dementia
Year: 2022
Evaluation of Plasma Biomarkers for A/T/N Classification of Alzheimer Disease Among Adults of Caribbean Hispanic Ethnicity
Journal: JAMA Network Open
Year: 2023
Increased Production of Pathogenic, Airborne Fungal Spores upon Exposure of a Soil Mycobiota to Chlorinated Aromatic Hydrocarbon Pollutants
Journal: Microbiology Spectrum
Year: 2023
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


