What is ChIP-seq
ChIP-sequencing (also known as ChIP-seq), which combines chromatin immunoprecipitation (ChIP) assays with DNA sequencing, is a powerful technique for genome-wide profiling of DNA-binding proteins, histone modifications or nucleosomes. ChIP is a type of immunoprecipitation (IP) experimental method used to isolate specific DNA sites in direct physical interaction with transcription factors and other proteins. In ChIP, specific antibodies are used to enrich DNA fragments bound by particular proteins or nucleosomes.
ChIP-seq was one of the early applications of NGS (next-generation sequencing), and the first study of large-scale profiling of the genome-wide histone methylations using ChIP-seq was published in 2007 (Barski et al., 2007). The sequencing of this study was performed on the platform of Solexa 1G genome analyzer. In the same year, Johnson et al. (2007) used ChIP-seq to generate the genome-wide mapping of transcription factor binding sites. Robertson et al. (2007) developed ChIP-seq to identify mammalian DNA sequences bound by transcription factors in vivo. These two papers also demonstrated the increased sensitivity and specificity of ChIP-seq. Owing to the rapid progress of NGS technology and the decreasing cost of sequencing, ChIP-seq has become an indispensable tool for characterization of epigenomes and gene regulation study (Park, 2009).
Difference between ChIp-chip and ChIp-seq
ChIP-chip, ChIP coupled with microarrays, and ChIP-seq are two standard techniques for identification of the genome wide DNA-proteins binding interactions. Taking advantage of sequencing technologies, ChIP-seq offers many advantages over ChIP-chip, as summarized in Table 1 (Park, 2009; Schones and Zhao, 2008).
Table 1. Comparison of ChIP-chip and ChIP-seq.
|
ChIP-chip |
ChIP-seq |
| Maximum resolution |
Array-specific, generally 30-100 bp |
Single nucleotide |
| Coverage |
Limited by sequences on the array; repetitive regions are usually masked out |
Limited only by alignability of reads to the genome; increases with read length; many repetitive regions can be covered |
| Flexibility |
Dependent on available products; multiple arrays may be needed for large genomes |
Genome-wide assay of any sequenced organism |
| Source of platform noise |
Cross-hybridization between probes and nonspecific targets |
Some GC bias can be present |
| Experimental design |
Single- or double-channel, depending on the platform |
Single channel |
| Cost-effective cases |
Profiling of selected regions; when a large fraction of the genome is enriched for the modification or protein of interest (broad binding) |
Large genomes; when a small fraction of the genome is enriched for the modification or protein of interest (sharp binding) |
| Required amount of ChIP DNA |
High (a few micrograms) |
Low (10-50 ng) |
| Dynamic range |
Lower detection limit; saturation at high signal |
Not limited |
| Amplification |
More required |
Less required; single-molecule sequencing without amplification is available |
| Multiplexing |
Not possible |
Possible |
How does ChIP-seq Work
The workflow of ChIP-seq used to profile the specific DNA binding sites for transcription factors, DNA-binding enzymes or other DNA-associated proteins (non-histone ChIP) and DNA sites correspond to modified nucleosomes (histone ChIP) is illustrated in Figure 1 (Park, 2009). Following ChIP protocols, the chromatin is fragmented and crosslinked proteins or modified nucleosomes immunoprecipitated using an antibody specific to the protein or the histone modification. After DNA purification and library construction, DNA fragments can be sequenced simultaneously on any of the sequencing platforms, such as Illumina Solexa Genome Analyzer, Roche 454 and Applied Biosystems (ABI) SOLiD platforms, and HeliScope by Helicos, as illustrated in Figure 1. With the tremendous progress of NGS technology, the Illumina platform, such as Hiseq, has been the most widely used platform for sequencing.
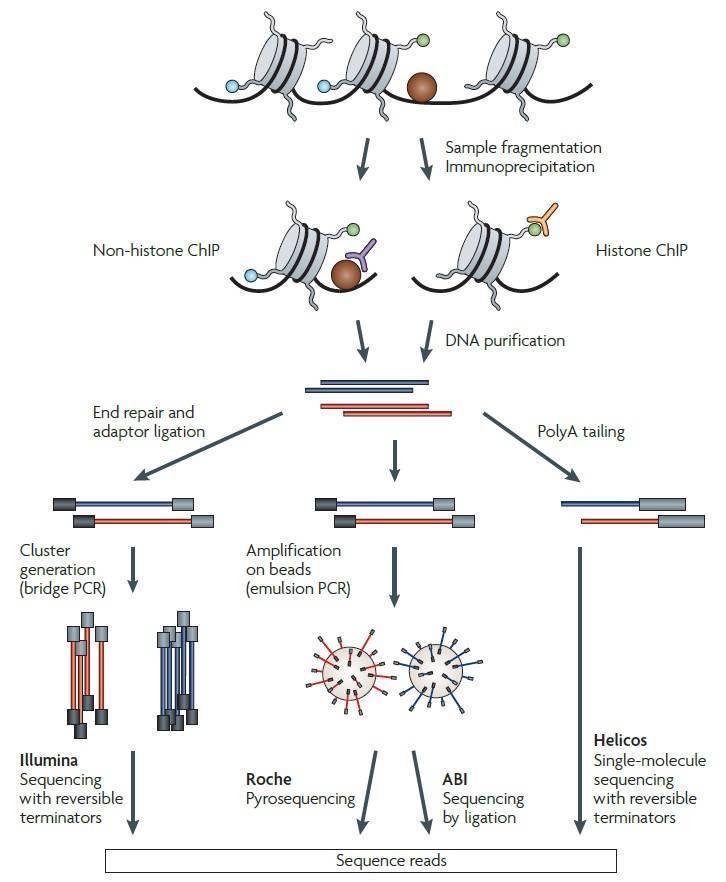 Figure 1. Overview of a ChIP-seq experiment (Park, 2009).
Figure 1. Overview of a ChIP-seq experiment (Park, 2009).
Experimental Design of ChIP-seq
The Encyclopedia of DNA Elements (ENCODE) and model organism ENCODE (modENCODE) consortia have developed a set of working standards and guidelines for ChIP-seq experiments based on experience of hundreds of ChIP-seq experiments (Landt et al., 2012). To obtain high-quality ChIP-seq data, there are several technical aspects should be considered in the ChIP-seq experimental design, including antibodies, cell number, controls, replicates, chromatin fragmentation, library construction and sequencing (Kidder et al., 2011).
Antibodies
The quality of antibodies used for ChIP is one of the most important factors that contribute to the quality of ChIP-seq data.
A sensitive and specific antibody will give a high level of enrichment. Limited efficiency of antibody is the main reason for failed ChIP-seq experiments.
Antibody validation and characterization should be done before the ChIP begin.
Cell number
As the signal-to-noise ratio (SNR) is directly correlated with the cell number, using the correct number of cells can help to diminish the background noise.
The abundance of the protein or histone modification to be investigated and the quality of the antibody should be considered when determining the number of cells.
Controls
An important part of ChIP-seq experimental design is determining which controls to use. A ChIP-seq peak should be compared with the same region in a matched control.
There are several different control types but no consensus on which is the most appropriate:
Input DNA.
Mock IP: DNA obtained from IP without antibody.
Nonspecific IP: using an antibody against a protein that is not known to be involved in DNA binding.
Replicates
High-quality ChIP-seq data sets are valuable resources for the community. Many factors, including cell-culture conditions, ChIP and library construction, may contribute to variability between data sets.
To ensure reliability of the data, biological replicate experiments are necessary.
Chromatin fragmentation
Before ChIP, chromatin must be fragmented into a manageable size.
ChIP-seq for DNA-binding proteins uses endonuclease digestion or sonication to fragment DNA.
ChIP-seq for histone modifications uses micrococcal nuclease (MNase) digestion to fragment DNA.
Library construction and sequencing
Libraries may be constructed from ChIP DNA by standard protocols specific to the sequencing platform.
Process in library construction and sequencing, including size selection, gel purification, PCR, single-end or paired-end sequencing strategy and sequencing depth, would affect the ChIP-seq data quality.
Considering the above technical aspects, a ChIP-seq experimental design that would obtain high-quality data is illustrated in Figure 2 (Kidder et al., 2011). At first, the appropriate controls for antibody specificity should be determined before ChIP. Chromatin is sheared into an ideal size range by sonication or enzymatic means after isolation of the ideal number of cells. Next, high-quality antibodies are used for ChIP. After purification of ChIP-enriched DNA, a library is constructed to allow sequencing on NGS platforms.
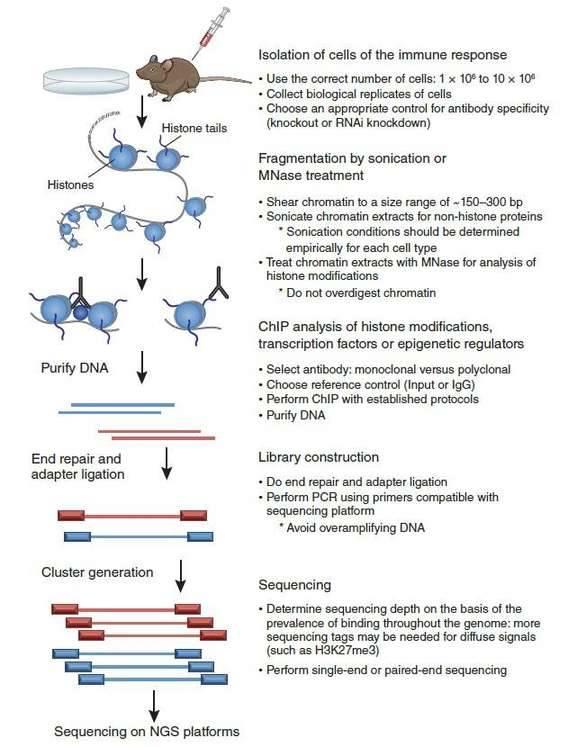 Figure 2. ChIP-seq experimental design (Kidder et al., 2011).
Figure 2. ChIP-seq experimental design (Kidder et al., 2011).
At CD Genomics, we provide you with high-quality sequencing and integrated bioinformatics analysis for your ChIP-Seq project, enabling accurately screen and determine the protein binding sites in the whole genome. If you have additional requirements or questions, please feel free to contact us.
Additional reading:
Pipeline and Tools for ChIP-seq Analysis
References:
- Barski, A., Cuddapah, S., Cui, K., Roh, T.Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I., and Zhao, K. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823-837.
- Johnson, D.S., Mortazavi, A., Myers, R.M., and Wold, B. (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497-1502.
- Kidder, B.L., Hu, G., and Zhao, K. (2011). ChIP-Seq: technical considerations for obtaining high-quality data. Nature immunology 12, 918-922.
- Landt, S.G., Marinov, G.K., Kundaje, A., Kheradpour, P., Pauli, F., Batzoglou, S., Bernstein, B.E., Bickel, P., Brown, J.B., Cayting, P., et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome research 22, 1813-1831.
- Park, P.J. (2009). ChIP-seq: advantages and challenges of a maturing technology. Nature reviews Genetics 10, 669-680.
- Robertson, G., Hirst, M., Bainbridge, M., Bilenky, M., Zhao, Y., Zeng, T., Euskirchen, G., Bernier, B., Varhol, R., Delaney, A., et al. (2007). Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature methods 4, 651-657.
- Schones, D.E., and Zhao, K. (2008). Genome-wide approaches to studying chromatin modifications. Nature reviews Genetics 9, 179-191.

 Sample Submission Guidelines
Sample Submission Guidelines


