
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

A large-scale F2 population can be utilized to perform Bulk Segregant Analysis (BSA) for initial mapping. Subsequently, within the identified mapping intervals, single nucleotide polymorphism (SNP) markers can be employed to construct detailed local linkage maps and conduct quantitative trait locus (QTL) mapping. This approach enhances the precision in locating target trait loci. Furthermore, integrating transcriptome sequencing data for joint analysis facilitates accelerated identification of candidate genes. This methodology is anticipated to dominate as the leading strategy for fine-mapping in the foreseeable future.
BSA, also known as mixed pool analysis or segregating population pool analysis, is a methodological approach that involves selecting individuals with extreme or representative phenotypes from a population to create mixed pools for analysis. By examining the differences in allele or molecular marker frequencies between these mixed pools, loci associated with specific traits can be mapped on the genome.
In comparison to traditional genetic research methods, a notable advantage of BSA is that it eliminates the need for genotyping all individuals within a population. Instead, it focuses on analyzing pooled samples from selected individuals based on their phenotypic characteristics. This approach significantly reduces the workload and costs associated with the study. For example, in a population of 500 individuals, genotyping of only two parental pools and two offspring pools may be sufficient to complete a BSA analysis (and even just the offspring pools alone can facilitate BSA analysis in the absence of parental pools). Furthermore, provided that the experimental design is sound, the statistical reliability of BSA analysis is comparable to methods involving the study of all individual samples.
Moreover, BSA is not exclusively reliant on sequencing technologies. It can be employed using molecular markers, microarrays, and high-throughput sequencing methods across various biological layers, including DNA, RNA, and proteins. However, as of now, comprehensive BSA methodologies at the protein level have not been fully established. Consequently, BSA analyses are predominantly concentrated on DNA and RNA layers.
For BSA, ensuring uniformity within the pool is crucial. This uniformity implies that the DNA sequenced should be evenly sourced from each individual. Therefore, it is recommended to first extract nucleic acids from each individual and then combine these extracts to form the pool, rather than mixing the samples prior to nucleic acid extraction.
The size of the pool and the sequencing depth are both critical factors that influence the precision of locus mapping. An increase in either pool size or sequencing depth generally enhances the accuracy of localization. However, beyond a certain threshold, further increases in pool size and sequencing depth result in diminishing returns with respect to improved localization accuracy. For conventional F2 populations, it is typically necessary to include more than 30 individuals per pool, with a sequencing depth of 20× for the parental sequences and 1× per individual for the offspring pools.
Given a fixed population size, the size of the pool must be appropriately limited to ensure that the pool accurately represents specific extreme traits. If the selected traits are not sufficiently extreme, this may lead to unsuccessful localization. To improve localization accuracy by increasing pool size, it is necessary to concurrently increase the overall population size. It is imperative to avoid mixing individuals from different populations with similar phenotypes for BSA analysis.
Population Construction: F1, F2, and BC1F1 populations are established.
Quantitative Trait Locus (QTL) Mapping: QTLs are mapped in the F2 population. Validation of QTL mapping results is performed using F2:3 progeny lines.
QTL Segregation: Molecular markers are utilized to select target QTLs that are heterozygous. BC1F1 single plants are identified where other QTLs are homozygous (a sufficient number of BC1F1 individuals is required). Subsequent self-pollination yields BC1F1:2 progeny lines.
Extreme Phenotype Selection: Two extreme phenotype groups are selected from the BC1F1:2 population.
Extreme Phenotype Pooling: DNA is extracted from the selected extreme phenotype groups and pooled to form two extreme pools.
Sequencing and Variant Calling: Sequencing is performed on the two pooled samples. Variants are identified by aligning the sequencing data against a reference genome.
Association Analysis: SNP-index analysis is conducted (Takagi H, et al., 2013; Mansfeld BN, et al., 2018).
For additional information on the workflow, refer to the "BSA-seq Technology Workflow"
You may interested in
A maize population study was conducted to identify QTLs associated with plant height. Using four generations of maize materials, a segregating F2 population was created from two inbred lines, HZS and 1462, which exhibit significant height differences. The range of individual plant heights in the F2 population varied from 199 cm to 307 cm, indicating a highly segregating trait, suggesting the involvement of multiple QTLs in controlling plant height.
Methodology
Population Construction:
The F2 segregating population was developed from the two inbred lines HZS and 1462.
QTL Mapping:
A total of 1,028 polymorphic markers were utilized to identify major-effect QTLs. Four QTLs were mapped to chromosomes 1, 3, 6, and 7, with phenotypic variation explained (PVE) ranging between 7% and 17%. The QTL mapping results were validated using F2:3 progeny lines.
Fine Mapping of qPH7:
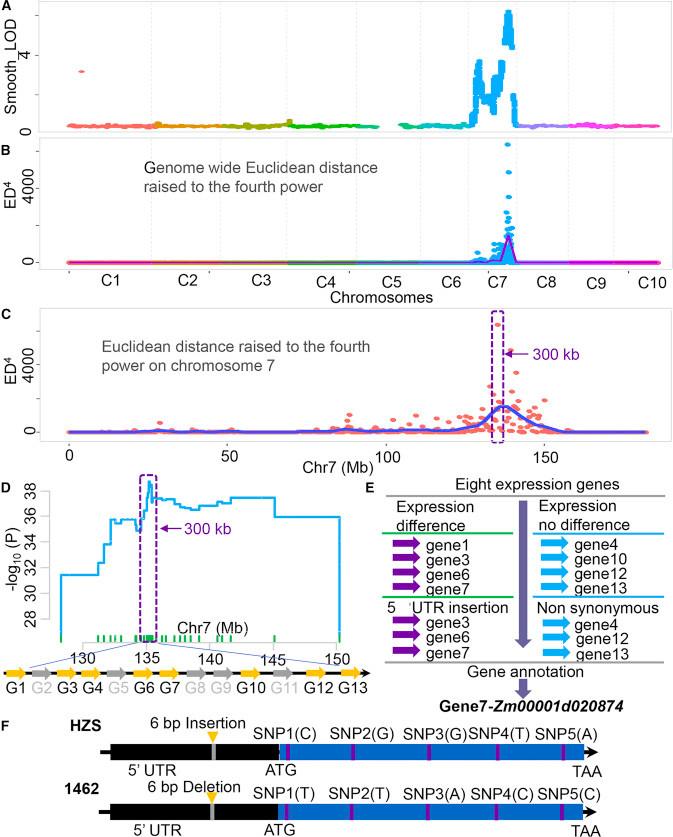
The QTL located on chromosome 7, designated as qPH7, was subjected to fine mapping using a QTL-seq strategy. From 813 BC1F1 single plants (heterozygous for qPH7 and homozygous for the other three QTL loci), 15 plants were selected, and BC1F1:2 progeny lines were generated. Plant height was assessed, resulting in 3,120 BC1F1:2 single plants. These were divided into two extreme phenotype groups: low-value (580 plants) and high-value (567 plants). DNA from these groups was pooled into low and high pools, respectively.
Genome-Wide Sequencing:
Whole-genome sequencing was performed on the pooled samples (coverage > 280×), detecting 197,021 high-quality SNPs. Marker genotyping confirmed the peak location between 135.1 and 135.2 Mb. Analysis using the smoothLOD and ED4 algorithms revealed a single peak on chromosome 7, located at 135.3 Mb.
Candidate Gene Identification
To further identify candidate genes, RNA-seq analysis was performed. Differential expression analysis showed that Zm00001d020874 exhibited significant differential expression between the shoot apical meristem (SAM) and juvenile internode tissues of the parent lines. Based on functional homologs in Arabidopsis and the smoothLOD mapping signal, Zm00001d020874 is considered a candidate gene for qPH7. Subsequent experiments, including CRISPR gene knockout and protein interaction assays, support the role of Zm00001d020874 as a gene controlling plant height.
Conclusion
The study demonstrates an effective approach for rapidly mapping QTLs associated with maize plant height and identifying candidate genes through a combination of QTL-seq and RNA-seq analyses. The findings contribute to a better understanding of the genetic control of plant height and provide a basis for further functional validation.
Technical Challenges in Mutant-Wild Type Hybrid Mapping
During the process of mapping intervals using mutants and wild-type hybrids, certain technical challenges may arise. Consequently, this approach should be used cautiously or avoided when possible due to these complexities.
Considerations in BSA and Candidate Gene Mapping
When applying BSA or other precision techniques to locate candidate genes, it is essential to note that the absence of polymorphism between the parental lines in a specific region does not necessarily indicate a false positive in the mapped interval. The molecular regulatory mechanisms underlying phenotypic variation between species are intricate and multifaceted. Such variation may be attributed to several factors, including SNPs, InDels, exon mutations, or variations in promoter regions.
It is a common challenge in gene mapping and cloning studies to encounter scenarios where the expected polymorphisms are not evident. However, there are instances where DNA methylation serves as a critical regulatory mechanism. For example, during the process of vernalization, low temperatures induce changes in DNA methylation states, which in turn regulate the expression of the FLOWERING LOCUS C (FLC) gene, ultimately affecting the flowering process in plants (Sheldon et al., 2000; Berry et al., 2015).
CD Genomics, as a leading provider of population genetic analysis and genotyping services, is committed to delivering exceptional and cost-effective solutions through a range of highly specialized technical methods. In the domain of population genetic analysis, the company excels in employing advanced techniques such as simplified genome sequencing, genome resequencing, BSA sequencing, Target-seq, and eukaryotic transcriptome sequencing. These methods are meticulously designed to meet the precise and efficient genetic analysis needs of a diverse clientele.
In the field of genotyping, CD Genomics demonstrates substantial expertise and technical proficiency. The company focuses on the development of comprehensive whole-genome SNP marker panels to provide clients with accurate and extensive genetic information. Additionally, CD Genomics offers high-throughput SNP genotyping services to accommodate large-scale sample processing requirements. The company is also proficient in utilizing SSR markers, including PAGE gel electrophoresis and agarose gel electrophoresis techniques, for genotyping analysis. This expertise is particularly valuable in the realm of crop variety fingerprinting, supporting agricultural genetic breeding and intellectual property protection.
Reference:


