The Importance of Plant Genetics and Gene Mapping
Plant genetics delves into the intricate structure, functional dynamics, and inheritance mechanisms of plant genes, directly impacting traits such as morphology and agronomic performance. This field of study provides foundational insights necessary for effective crop breeding and enriches our understanding of genetic diversity and plant adaptability to diverse environmental conditions. Gene mapping emerges as an essential tool within plant genetics, facilitating the precise linkage of specific traits to genomic loci. This methodology is instrumental in identifying genes or quantitative trait loci (QTLs) associated with crucial agronomic traits. The development of high-density genetic maps plays a pivotal role in enhancing breeding efficiency and supporting the creation of novel plant varieties.
The Distinctive Value of Bulk Segregant Analysis in Plant Research
Bulk Segregant Analysis (BSA) stands out as a robust gene mapping technique, adept at swiftly localizing genes or QTLs related to target traits through the genotyping of individuals displaying extreme phenotypes from an F2 population. Renowned for its efficiency and accuracy, BSA is particularly advantageous for dissecting complex traits. It is capable of producing significant findings from a limited number of samples. The integration of BSA with high-throughput sequencing technologies, such as BSA-Seq, further augments its resolution and analytical power. BSA has been successfully employed to pinpoint genes influencing traits like grain number in rice and seed color in rapeseed. Additionally, it provides critical insights into plant responses to environmental stress and disease resistance, offering valuable contributions to crop breeding strategies and enhancing global food security.
Case Studies of Bulk Segregant Analysis in Plant Genetics
Case 1: Mapping disease resistance genes using Bulk Segregant Analysis
Bulked segregant analysis is frequently used to find molecular markers linked to traits of interest, such as disease resistance. BSA involves creating groups of plants with opposing phenotypes. Recent research has used bulked segregant RNA-Seq (BSR-Seq), which combines BSA and RNA-Seq techniques, to map genes of interest. For example, BSR-Seq of genome-wide DNA variations in a B. rapa population was used to map the clubroot resistance gene Rcr1. In another instance, resistance to pathotype 5X was mapped to the same region as Rcr7. It was discovered that the B. oleracea line 'Kilaherb' was the only one that carried the SNP alleles linked to Rcr7. Furthermore, a study utilized next-generation sequencing-based BSA to identify significant QTLs associated with Northern Corn Leaf Blight (NCLB) in Zea mays. The study identified 10 QTLs on chromosomes 1, 2, 3, and 5, along with 27 candidate genes related to disease resistance, including AATP1 and STICHEL-like 2.
Case 2: Studying plant growth traits with Bulk Segregant Analysis
BSA can be used to study plant growth characteristics. BSA has been used to identify genes that control plant height and, as a result, the dwarfing trait in castor beans. The gene Rc5NG4-1 was discovered to be responsible for controlling plant height. In another study, BSA-Seq was used to identify 13 candidate regions linked to the temperature-sensitive albino phenotype of B. napus. RNA-Seq analysis revealed four genes, including those encoding TOC75-3 and TIC62, as the most likely candidates involved in chloroplast development. BSA has also been applied to improve drought resistance in maize. Molecular markers showing polymorphism between parents and closely linked to major QTLs regulating specific traits co-segregate with that QTL.
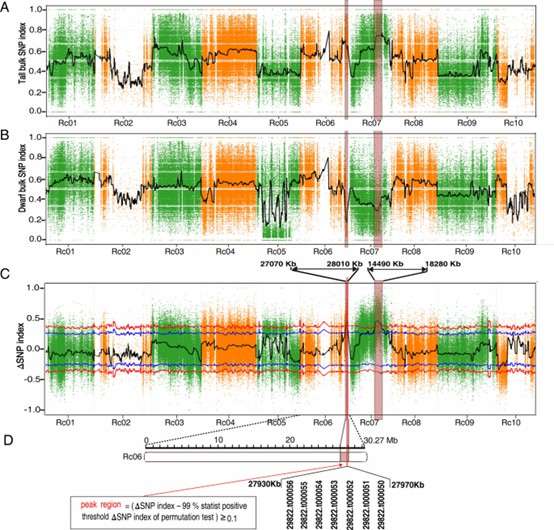 BSA for QTLs contributed to plant height using 1 Mb sliding windows with a step size of 10 Kb. (Wang, Z., et al., 2021)
BSA for QTLs contributed to plant height using 1 Mb sliding windows with a step size of 10 Kb. (Wang, Z., et al., 2021)
Services you may interested in
Want to know more about the details of Bulk Segregant Analysis? Check out these articles:
Bulk Segregant Analysis in Plant Breeding
Bulk Segregant Analysis plays a pivotal role in plant breeding by rapidly localizing genes or QTL associated with target traits through phenotypic segregation. This method accelerates the breeding process and enhances efficiency.
The Role of BSA in Plant Breeding
BSA is a gene mapping technique based on population genetics. It involves constructing a segregation population in the F2 generation, selecting individuals with extreme phenotypes (forming bulks), and analyzing their genotypes to swiftly identify genetic markers or QTL related to the target traits. The method offers several advantages:
- Efficiency: BSA significantly reduces the number of experimental samples required, avoiding the high costs and inefficiencies associated with genotyping each plant individually.
- Versatility: It is applicable to both self-pollinating and cross-pollinating crops and can handle large quantities of segregating material.
- Accuracy: By statistically analyzing the genotypes of individuals with extreme phenotypes, BSA can more precisely locate QTL associated with specific traits.
In studies of cold tolerance in rice, BSA combined with whole-genome resequencing and QTL-seq analysis has successfully identified candidate genes for cold resistance. For instance, a study constructed a backcross recombinant inbred line (BRIL) population to identify QTLs associated with cold tolerance. The researchers discovered 73 QTLs linked to various traits under cold stress conditions, including survival rate and plant height. Notably, candidate genes such as CBF/DREB and MYB were identified, providing insights into the genetic mechanisms underlying cold tolerance in rice.
Additionally, BSA has been instrumental in mapping dwarfing genes in apples. By utilizing BSA, researchers were able to pinpoint key genes responsible for dwarfing traits, which are crucial for improving apple tree architecture and fruit production efficiency. For example, a comprehensive BSA analysis revealed several candidate genes that may control plant height in various species, including those responsible for the dwarfing trait in castor bean.
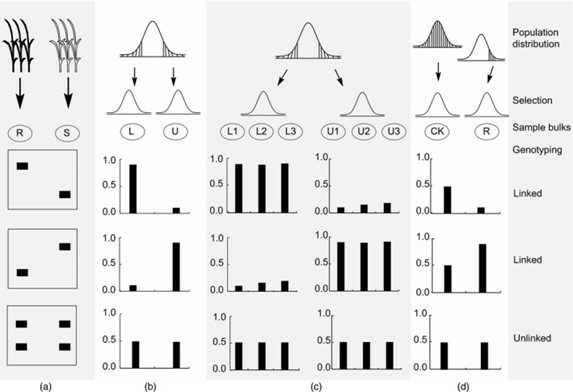 Four types of bulked sample analysis (BSA). (Zou, et al., 2016)
Four types of bulked sample analysis (BSA). (Zou, et al., 2016)
Furthermore, BSA has facilitated the development of molecular markers for seedlessness in grapes. A study focused on identifying markers linked to the seedless trait through BSA, allowing breeders to exclude seeded segregants early in the breeding process. This approach not only accelerates the breeding timeline but also reduces costs associated with long-term plant development. The identification of SNP markers associated with the VviAGL11 gene has been particularly significant in understanding the genetic basis of seedlessness in table grape.
Enhancing Breeding Efficiency
BSA technology can further boost plant breeding efficiency through various approaches:
- Integration with High-Throughput Sequencing: Traditional BSA relies on molecular markers (e.g., SSR, SNP) for gene mapping, whereas BSA-seq (BSA combined with high-throughput sequencing) enables rapid identification of genes or QTL associated with traits directly from a genomic perspective. For instance, the OcBSA tool uses NGS technology to efficiently analyze segregating F1 populations of cross-pollinated species, significantly enhancing gene discovery efficiency.
- Optimized Experimental Design: Improving BSA experimental design, such as increasing the sample size or conducting multiple replicated experiments, can enhance the reliability and resolution of results.
- Combination with Other Molecular Marker Techniques: Utilizing RNA-seq in BSA analysis can reveal regulatory genes for specific traits, such as stigma development during pollination.
- Development of Specialized Software Tools: Tools like the QTLseqr and PyBSA R packages provide more efficient statistical analysis methods for BSA.
BSA-seq technology has significantly advanced plant breeding by identifying key genes affecting various traits. For example, it has successfully pinpointed genes responsible for seed color in rapeseed (Brassica napus), such as BnaPAP2.C6a and BnaPAP2.A7b, which are linked to purple stems and red flowers, respectively. This research enhances anthocyanin inheritance understanding and provides valuable genetic resources for breeding programs.
BSA has also been crucial in identifying molecular markers for smut disease resistance in sugarcane. A study using BSA-seq identified several QTLs linked to resistance to Sporisorium scitamineum, which can be integrated into breeding programs to enhance sugarcane resilienc.
Additionally, BSA has improved yellow seed color in Brassica juncea by identifying candidate genes like BjuA09PAL2, associated with yellow seed coloration. This discovery is important for breeding yellow-seeded varieties, which offer agronomic advantage.
Bulk Segregant Analysis in Plant Genomics
The fusion of BSA with Next-Generation Sequencing (NGS) has emerged as a formidable tool in plant genomics, enabling the efficient and precise localization of genes or QTL associated with specific traits. Beyond gene mapping, this methodology holds substantial potential for plant genome editing applications, particularly in gene function validation and the analysis of polygenic traits. Nonetheless, its application faces challenges related to data processing complexity and costs, necessitating further technical refinement and resource investment.
Applications of BSA Combined with NGS in Plant Genomics
BSA is a conventional genetic analysis technique that swiftly pinpoints QTL controlling specific traits by segregating extreme individuals from an F2 population. The advent of NGS has significantly enhanced the efficiency and precision of gene mapping when combined with BSA. For instance:
- QTL-seq Method: This approach involves whole-genome resequencing of populations segregated via BSA, combined with statistical models such as the G Prime method, to rapidly pinpoint trait-associated QTL. It has been extensively applied across various crops, including rice, wheat, and tomato.
- BSA-seq Technology: By employing high-throughput sequencing on BSA-separated populations, researchers can swiftly identify SNP markers and candidate genes related to traits. For example, in tomato, BSA-seq successfully localized a gene controlling stigma exertion.
- RNA-BSA: By integrating RNA sequencing (RNA-seq) technologies, RNA-BSA allows for the exploration of gene expression differences and their correlation with traits. In foxtail millet, BSA-Seq combined with RNA-Seq identified candidate genes related to inflorescence development.
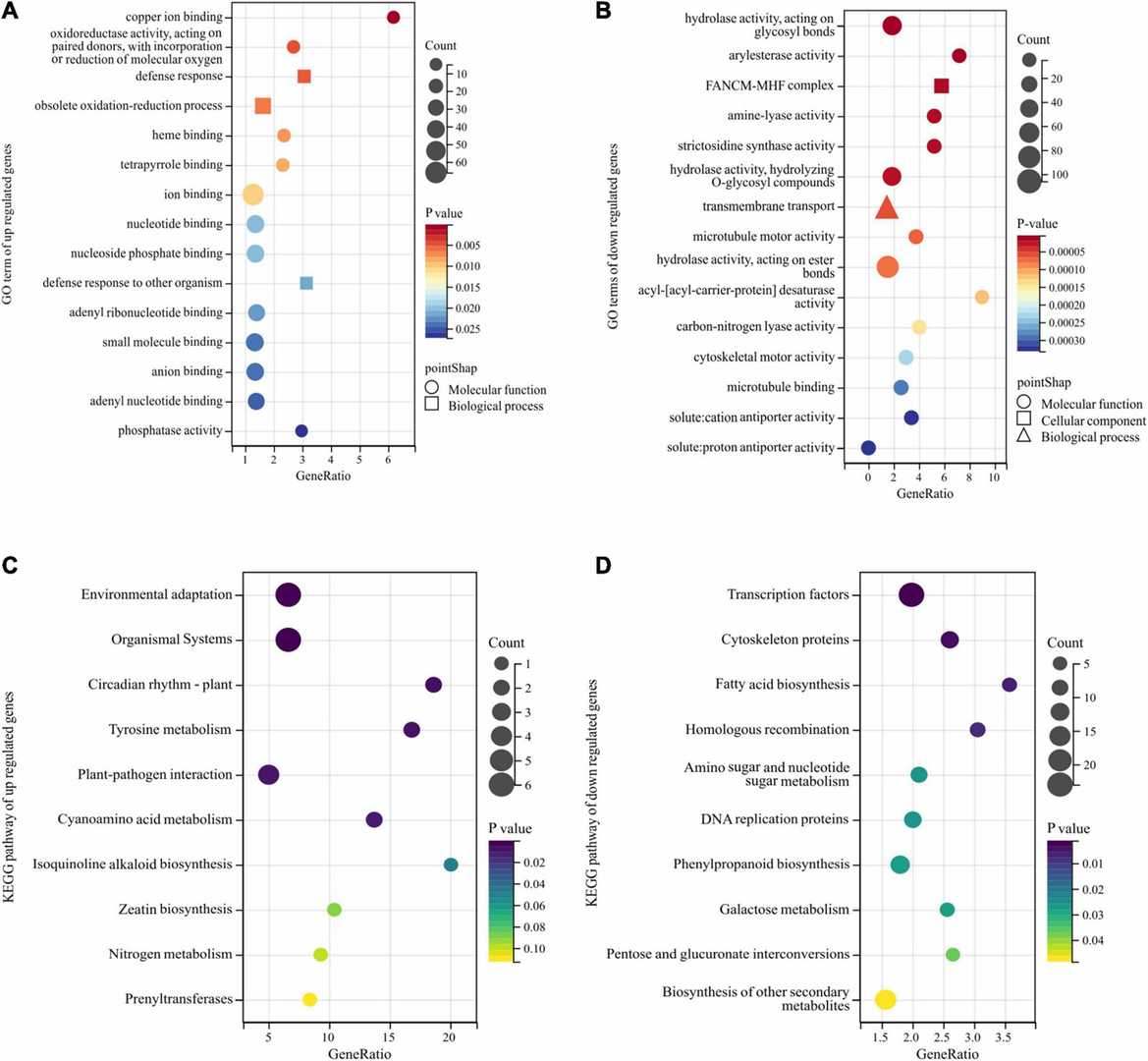 Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEGs obtained by bulk segregant RNA sequencing (BSR-Seq). (Gao, Yongbin, et al., 2022)
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of DEGs obtained by bulk segregant RNA sequencing (BSR-Seq). (Gao, Yongbin, et al., 2022)
Potential Applications of BSA in Plant Genome Editing
The amalgamation of BSA with NGS is not restricted to gene mapping but also extends critical support to plant genome editing:
- Gene Function Validation: Populations isolated through BSA can facilitate the rapid validation of gene editing tools like CRISPR/Cas9. In cabbage, for example, BSA segregation and resequencing successfully identified QTL associated with disease resistance.
- Polygenic Trait Analysis: Many plant traits are governed by multiple genes. The integration of BSA with NGS can unveil the genetic mechanisms underlying these polygenic traits. In soybean, BSA-seq successfully mapped several QTL controlling seed size and weight.
- Gene Mapping in Cross-Pollinated Species: For cross-pollinated species such as cabbage and maize, traditional inbreeding methods can be time-consuming. The combination of BSA and NGS provides an efficient route to analyze the genomic structure and genetic control of important traits in these species.
Advantages and Challenges of BSA in Plant Research
Bulk Segregant Analysis is a widely utilized genetic analysis method in plant research. Its strengths and challenges can be examined from multiple perspectives.
Advantages:
- Rapidity: BSA efficiently screens for genes or QTL associated with target traits by segregating extreme phenotype individuals from an F2 generation and combining them into two pools (commonly referred to as "bulk" and "small bulk"). This approach circumvents the need for individual analysis of the entire population, significantly reducing research time.
- Efficiency: By using combined pools of individuals with extreme phenotypes, BSA greatly enhances the efficiency of target gene mapping. For instance, in the study of stigma exertion genes in tomatoes, BSA coupled with high-throughput sequencing successfully pinpointed the target gene.
- Cost-Effectiveness: Compared to traditional whole-genome sequencing or high-density SNP chips, BSA is relatively cost-effective. This is because BSA requires sequencing only the DNA of individuals with extreme phenotypes, rather than the entire population.
Challenges:
- Challenges in Sample Selection: BSA relies on accurately choosing and segregating individuals with extreme phenotypes. If sample selection is inaccurate, it may lead to biased results. For example, imprecise sample selection in some studies might result in inaccurate localization of target genes.
- Complexity of Data Analysis: With the advancement of high-throughput sequencing technologies, the data analysis in BSA has become increasingly complex. Techniques like BSA-seq, which integrate traditional BSA strategies with high-throughput sequencing, enhance precision but also complicate data analysis.
- False Positive and False Negative Rates: Since BSA only analyzes individuals with extreme phenotypes, it may introduce false positive or false negative outcomes. In some scenarios, extreme phenotypes may not be solely determined by the target gene but may be influenced by other genetic or environmental factors.
In sum, while BSA offers a rapid, efficient, and cost-effective method for genetic analysis in plant research, careful attention must be paid to the challenges of sample selection, data complexity, and potential biases in results to fully harness its capabilities.
Conclusion
BSA is a powerful tool in plant research, offering rapid, efficient, and cost-effective methods for gene mapping and QTL identification. By leveraging extreme phenotypes and integrating with next-generation sequencing technologies, BSA accelerates the discovery of genes associated with important traits, enhancing breeding efficiency and supporting the development of new plant varieties. Despite challenges in sample selection and data complexity, BSA remains a valuable technique for advancing plant genetics.
Looking ahead, the future of plant genetics research is promising. Continued advancements in sequencing technologies and bioinformatics will further enhance the precision and applicability of BSA. The integration of BSA with other molecular techniques and the development of specialized software tools will streamline data analysis and improve the reliability of results. As plant genetics research progresses, BSA will play a crucial role in uncovering the genetic basis of complex traits, contributing to global food security and sustainable agriculture.
References:
- Dakouri, A., Zhang, X., Peng, G. et al. Analysis of genome-wide variants through bulked segregant RNA sequencing reveals a major gene for resistance to Plasmodiophora brassicae in Brassica oleracea. Sci Rep 8, 17657 (2018). https://doi.org/10.1038/s41598-018-36187-5
- Zhai, Ruining, et al. "SNP-based bulk segregant analysis revealed disease resistance QTLs associated with northern corn leaf blight in maize." Frontiers in Genetics 13 (2022): 1038948. https://doi.org/10.3389/fgene.2022.1038948
- Wang, Z., Yu, A., Li, F. et al. Bulked segregant analysis reveals candidate genes responsible for dwarf formation in woody oilseed crop castor bean. Sci Rep 11, 6277 (2021). https://doi.org/10.1038/s41598-021-85644-1
- Ye, Shenhua, et al. "Bulk segregant analysis-sequencing and RNA-Seq analyses reveal candidate genes associated with albino phenotype in Brassica napus." Frontiers in Plant Science 13 (2022): 994616. https://doi.org/10.3389/fpls.2022.994616
- Quarrie, Steve A., et al. "Bulk segregant analysis with molecular markers and its use for improving drought resistance in maize." Journal of experimental botany 50.337 (1999): 1299-1306. https://doi.org/10.1093/jxb/50.337.1299
- Royo, Carolina, et al. "The major origin of seedless grapes is associated with a missense mutation in the MADS-box gene VviAGL11." Plant physiology 177.3 (2018): 1234-1253. doi: 10.1104/pp.18.00259
- Ocarez, Nallatt, et al. "Unraveling the deep genetic architecture for seedlessness in grapevine and the development and validation of a new set of markers for VviAGL11-based gene-assisted selection." Genes 11.2 (2020): 151. doi: 10.3390/genes11020151
- Zou, Cheng, Pingxi Wang, and Yunbi Xu. "Bulked sample analysis in genetics, genomics and crop improvement." Plant biotechnology journal 14.10 (2016): 1941-1955. doi: 10.1111/pbi.12559
- Gao, Yongbin, et al. "Conjunctive analyses of bulk segregant analysis sequencing and bulk segregant RNA sequencing to identify candidate genes controlling spikelet sterility of foxtail millet." Frontiers in Plant Science 13 (2022): 842336. https://doi.org/10.3389/fpls.2022.842336
- Chen, Daozong, et al. "Fine mapping of genes controlling pigment accumulation in oilseed rape (Brassica napus L.)." Molecular Breeding 43.3 (2023): 19. doi: 10.1007/s11032-023-01365-5
- Wang, Yang, et al. "Identification of Yellow Seed Color Genes Using Bulked Segregant RNA Sequencing in Brassica juncea L." International Journal of Molecular Sciences 25.3 (2024): 1573. doi: 10.3390/ijms25031573

 Sample Submission Guidelines
Sample Submission Guidelines


