
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Contemporary life sciences are witnessing transformative advances in microbiome investigations, with amplicon sequencing emerging as a sophisticated analytical approach for comprehensively characterizing microbial ecological landscapes. This scholarly exposition critically examines two pivotal methodological frameworks in molecular microbial taxonomy: Operational Taxonomic Unit (OTU) clustering and Amplicon Sequence Variant (ASV) analysis. By meticulously exploring their technological foundations, historical trajectories, and contemporary research applications, we aim to provide researchers with a nuanced, evidence-based framework for methodological selection.
Amplicon sequencing, especially 16S rRNA sequencing, has become an important tool in microbiome research. Compared to whole-genome sequencing, it offers significant advantages, including lower cost, smaller sample size requirements, the ability to avoid host DNA contamination, and the ability to quickly obtain microbial taxonomic information.
However, despite the important role of amplicon sequencing in microbiome research, it still faces significant technical challenges, primarily stemming from random errors during the sequencing process. Specifically, these challenges include:
To address these issues, two commonly used analysis strategies—OTU and ASV—have been developed to reduce the impact of sequencing errors. The question then arises: in amplicon sequencing analysis, should one choose OTU or ASV?
Microbial community analysis traditionally employs the Operational Taxonomic Unit (OTU) clustering methodology, which categorizes genetic sequences based on precise similarity metrics. The fundamental approach revolves around aggregating genomic fragments that demonstrate significant sequence homology. The main features of this method include:
Similarity Threshold: Microbiological taxonomic protocols conventionally establish a 97% sequence similarity criterion for classification. Researchers consider this percentage a robust benchmark for identifying potentially congruent biological entities. Such a stringent threshold enables precise delineation of taxonomic boundaries while minimizing classification ambiguity.
Clustering Principle: The clustering mechanism prioritizes sequences with higher prevalence, strategically integrating low-frequency genetic fragments with more dominant representations. This computational approach presumed that abundant sequences more accurately reflect genuine biological signatures, thereby minimizing potential artifacts introduced by rare or potentially erroneous genetic variants.
Error Control: Rigorous quality control mechanisms are implemented to constrain intra-OTU genetic variability. By maintaining sequence divergence within a narrow 1% error margin, researchers can mitigate potential misclassifications arising from sequencing inaccuracies or technical variations.
The development of the OTU clustering method has been accompanied by the emergence of various algorithms, some of the representative ones include:
UPARSE (Robert C. Edgar, 2013): The UPARSE algorithm significantly improves the accuracy of amplicon sequencing studies by effectively removing sequencing errors and chimeras. It uses a greedy clustering strategy to ensure that the similarity between all paired OTU sequences is below 97%, with each OTU being the most abundant sequence in its neighborhood.
OneUniq: An optimization based on UPARSE, OneUniq further improves the credibility of OTUs. This algorithm enhances the recognition of low-abundance sequences by improving the processing workflow, thereby reducing false positive results.
Although the OTU clustering method has been widely applied in microbiome research, it also has some limitations:
Failure to Capture Subtle Sequence Variations: The OTU clustering method may fail to capture subtle variations in microbial communities because it relies on a fixed similarity threshold, which may result in some important information being overlooked.
SNPs Integrated into a Single OTU: In some cases, single nucleotide polymorphisms (SNPs) may be incorrectly integrated into the same OTU, which can affect the understanding of species diversity.
Subjectivity of Sequence Similarity Threshold: While using 97% as the similarity threshold is an industry standard, this choice is somewhat subjective. Different researchers may select different thresholds depending on specific circumstances, which can lead to inconsistencies in results.
The ASV (Amplicon Sequence Variant) analysis method represents a significant technological leap in high-precision microbiome analysis. It uses statistical models and algorithms to correct sequencing errors, revealing the true composition of microbial communities at single-base resolution. The ASV method significantly reduces noise introduced by clustering thresholds and sequencing errors in traditional clustering methods, providing more reliable data support for ecological models and functional predictions.
Key features of the ASV method:
| Feature | Description |
| Similarity Threshold | The similarity threshold for ASVs is 100%, avoiding the impact of manually set clustering thresholds (e.g., 97% or 99%) on the analysis results. |
| Analysis Strategy | The method employs statistical-based sequence error correction algorithms, which accurately identify and correct sequence variations by modeling sequencing errors. |
| Resolution | ASVs detect differences down to a single-base level, allowing the identification of more subtle biological variations in microbial communities. |
DADA2 (Divisive Amplicon Denoising Algorithm) is one of the core algorithms for ASV analysis, introduced by a team from Stanford University in 2016. Its design goal is to achieve error correction and precise variation detection in sequence data using statistical and machine learning models. The key technical features of DADA2 are as follows:
Statistical Learning of Variation Probabilities: DADA2 uses a Poisson distribution-based probability model to analyze each base position in the sequencing data, accurately calculating the likelihood of sequence variation. This method significantly improves the detection of low-abundance and rare variations, minimizing false positive results.
Divisive Clustering Algorithm: Through an iterative algorithm, DADA2 separates noise from real sequences, enabling the isolation of true sequence variations without relying on manually set similarity thresholds. This method overcomes the limitations of traditional OTU clustering that may overlook biological details.
Preservation of True Sequence Variations: The ASV sequences generated by DADA2 are consistently accurate, ensuring reproducibility and comparability across different studies. This method is particularly suited for high-resolution analytical scenarios in ecology, such as species distribution pattern studies and functional predictions.
The ASV analysis method offers significant advantages. Firstly, it provides higher resolution in microbial diversity analysis. Compared to the traditional OTU method, ASV can precisely differentiate the species composition of microbial communities at the single-base level, allowing for a more accurate capture of the distribution and dynamics of different microbial populations in the environment. Furthermore, through the statistical model in DADA2, ASV effectively removes false sequences caused by PCR amplification and sequencing errors, significantly improving the authenticity and reliability of the analysis results. This provides a stronger foundation for analyzing community diversity and ecological patterns. Additionally, the ASV method excels in detecting rare variations and low-abundance species, revealing the complexity of ecosystems and the diversity of microbial functions, while avoiding the loss of biological information due to manually set clustering thresholds. This makes ASV a key tool in microbial ecology research.
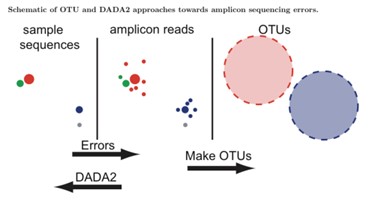 Figure 1.The difference between ASVs and OTUs(Callahan et al. 2016)
Figure 1.The difference between ASVs and OTUs(Callahan et al. 2016)
In microbiome research, selecting the appropriate analytical method is crucial for experimental design, data processing, and the scientific reliability of the final conclusions. Based on the type of study and technical requirements, the choice of method should carefully consider the specific needs of the research context as well as various technical factors, ensuring the accuracy and ecological relevance of the data analysis.
Different types of research and objectives determine the suitability of the ASV or OTU method. Below are practical recommendations based on typical research scenarios:
| Research Type | Recommended Method | Key Considerations |
| 16S rRNA Sequencing | ASV | More suitable for high-resolution analysis of short fragment regions, such as V4-V5 primer regions |
| Third-Generation Full-Length Amplicons | OTU | Better suited for long fragment sequence analysis, recommended to use a 98.5%-99% similarity threshold |
In practical applications, the choice of method is influenced by a range of technical and experimental design factors:
As microbiome research continues to advance, analytical methods and technologies are evolving rapidly, with trends toward diversification and increased precision. Looking ahead, both ASV and OTU methods are expected to align progressively in areas such as technical optimization, cross-platform standardization, and low-abundance sequence processing strategies, providing more reliable and flexible tools for scientific research.
With the development of deep learning and artificial intelligence technologies, machine learning algorithms are expected to deepen their applications in bioinformatics. For example, sequence error correction and classification models based on deep neural networks will enable more efficient handling of massive sequence datasets, enhancing the accuracy of microbiome analysis. Methods like DADA2 may further integrate machine learning techniques to dynamically predict the presence probability and functional contributions of low-abundance species. Currently, statistical-based sequence error correction tools like DADA2 may evolve into comprehensive analysis tools that combine multimodal data, such as transcriptomics and metabolomics.
Currently, significant differences in data quality, length, and noise characteristics exist between sequencing platforms like Illumina, PacBio, and Oxford Nanopore, making it difficult to compare results. In the future, developing a standardized analytical framework for cross-platform data will be a key direction in technological advancement. By establishing unified data formats, quality control standards, and analysis parameters, integrated analysis of multi-platform data will be facilitated, promoting global collaboration in microbiome research.
As sequencing depth increases and application scenarios become more complex, noise reduction algorithms will continue to evolve, especially in handling low signal-to-noise ratio and high-throughput data. For example, intelligent noise recognition models and dynamic threshold adjustment methods may be developed to address the challenges posed by high noise and heterogeneity in environmental samples. Innovative algorithms will further improve the sensitivity and accuracy of ASV while reducing computational resource consumption.
Low-abundance sequences play a crucial role in microbiome research, but their processing has long been challenging due to noise and false sequences. In recent years, algorithms have converged on a common approach for handling low-abundance sequences, forming a practical consensus:
DADA2 uses a strict statistical model to automatically remove singleton sequences (those with a frequency of 1) from samples. This strategy effectively avoids false positive sequences caused by amplification and sequencing errors, ensuring the accuracy of the analysis.
Amplicon sequencing analysis, as a core tool in microbiome research, does not have an absolute "best" method. Both the traditional OTU analysis and the emerging ASV analysis have their unique applicable scenarios and technical advantages. The key is for researchers to choose the most suitable method based on specific research objectives, experimental design, and sample characteristics, while balancing computational efficiency and analytical accuracy to retain the most accurate biological information.
In recent years, the ASV analysis method has gradually become the preferred choice for researchers due to its higher resolution, lower false sequence rate, and efficient capture of rare species and biological variation. The ASV method, relying on statistical models and refined sequence error correction techniques, provides a more accurate tool for revealing microbial community structure and ecological function. This method is particularly suitable for in-depth studies of complex samples, such as the dynamic changes in microbial communities in anaerobic digestion systems.
With its comprehensive sequencing platforms and bioinformatics analysis capabilities, CD Genomics offers professional support to researchers in amplicon sequencing and data mining processes. Whether using Illumina short-read platforms or long-read platforms like PacBio and Oxford Nanopore, CD Genomics integrates the latest algorithms (such as DADA2) and diverse analysis pipelines to help researchers achieve more insightful results. From experimental design to data analysis, the full-service workflow ensures high accuracy and reliability of research outcomes, opening up more possibilities for scientific exploration.
References:


