
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Our SMRT-based microbial epigenomics is capable of long-read lengths and enables mapping of methylation patterns within highly repetitive genomic regions. The SMRT sequencing technology can simultaneously determine nucleotide sequence and the methylation status of each nucleotide, through real-time observation of DNA polymerase kinetics.

We are dedicated to providing outstanding customer service and being reachable at all times.
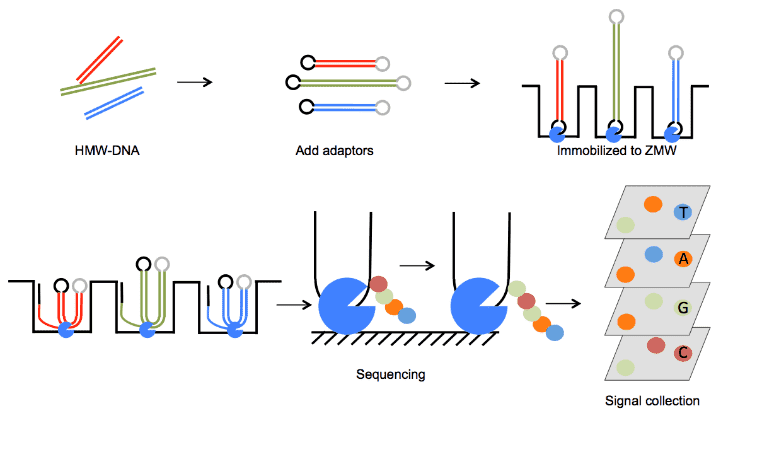
Microbial genomes contain a variety of base modifications, most of which are methylations occurring at adenine or cytosine residues. DNA methylation plays an important role in microbial defense against foreign DNA. During SMRT sequencing, DNA polymerases catalyze the incorporation of fluorescently labeled nucleotides into complementary nucleic acid strands. The arrival times and durations of the resulting fluorescence pulses yield information about polymerase kinetics and enable us to detect DNA modifications including 4-methylcytosine (m4C), 6-methyladenosine (m6A), 5-hydroxymethylcytosine (5hmC), and 5-methylcytosine (m5C), by measuring the variation in the polymerase kinetics. We also provide Nanopore-based epigenomics service for microbial epigenomics studies.
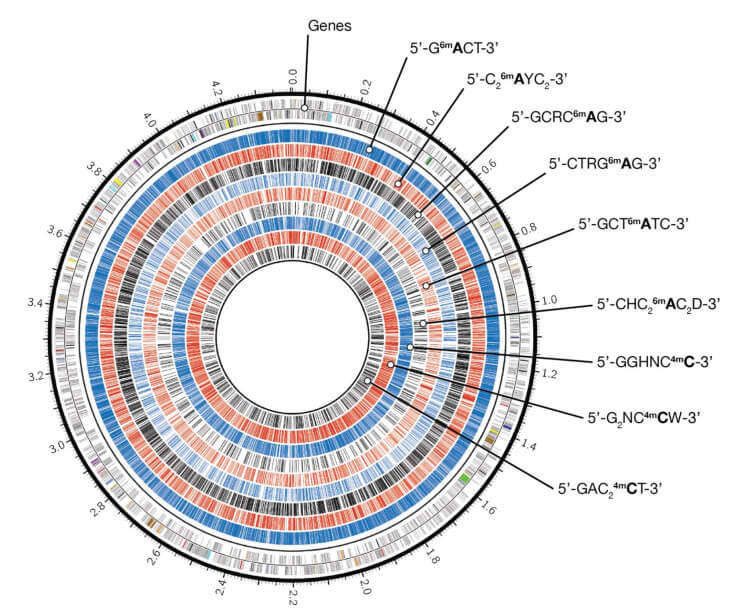
In addition to the detection of DNA methylation, PacBio SMRT sequencing also allows us to reveal phase variation of restriction-modification (R-M) genes that regulate batteries of genes involved in pathogenesis, host adaption, and antibiotic resistance. Not only driving the bacterial evolution by increasing C-T mutations and double-strand breaks, the R-M systems can also alter the transcription of genes involved in virulence, host adaption, pathogenicity, and antibiotic resistance. SMRT-based microbial epigenomics can be used to understand the pattern and function of DNA methylation, identify targets for therapeutic treatment, and reveal phase variation of R-M genes. It can be used to study cell cycle control and DNA repair, developmental biology, and cancer research.

Sampling kits: We provide a range of microbial sampling kits for clients, including MicroCollect™ oral sample microbial collection products and MicroCollect™ stool sample collection products.
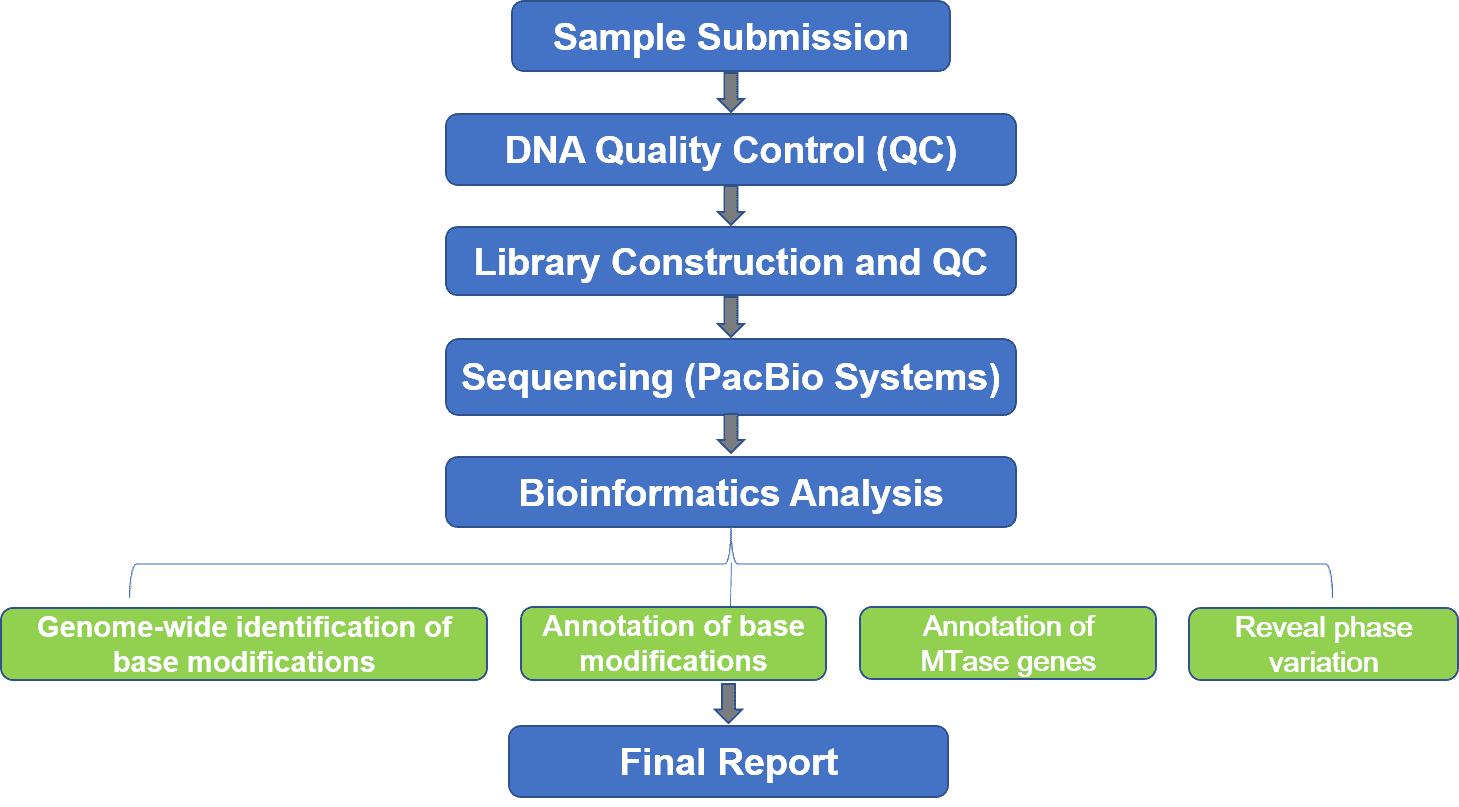
Deliverables: Raw data files in BAM format, demultiplex CCS reads in FASTQ format, quality-control dashboard, statistic data, and your designated bioinformatics analysis report.
References
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to info@cd-genomics.com for inquiries.
Please fill out the form below: ×


Follow us on: