Navigation
- Home
- Services
- Microbial Diversity Analysis – 16S/18S/ITS Sequencing
- Metagenomics
- Microbial Whole Genome Sequencing
- Microbial Identification
- Microbial Characterization
- Microbial Functional Gene Analysis
- Microbial Epigenomics
- Antibiotic Resistance Genes (ARGs) Analysis Solution
- Microbial PacBio SMRT Sequencing
- Microbial Nanopore Sequencing
- Microbial Transcriptomics
- Microbial Bioinformatics
- Other
- Microbial Metabolomics Analysis Service
- Solutions
- Microecology and Human/Animal Health
- Environmental Microbiology Solutions
- Agricultural Microbiology Solutions
- Pharmaceutical Microbiology Solutions
- Industrial Microbiology Solutions
- Microecology and Cancer Research Solutions
- Microecology and Biofilms
- Pathogen Sequencing Solutions
- Environmental DNA (eDNA) Analysis Solution
- Products
- Resource
- Company
- Sample Submission Guidelines


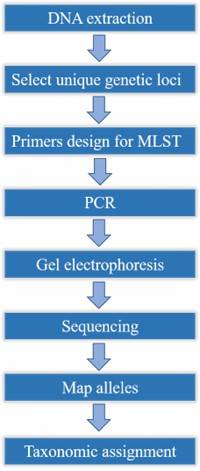
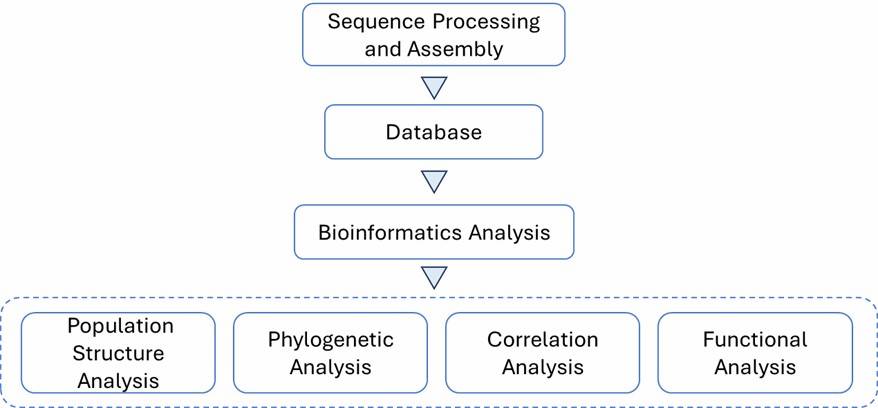
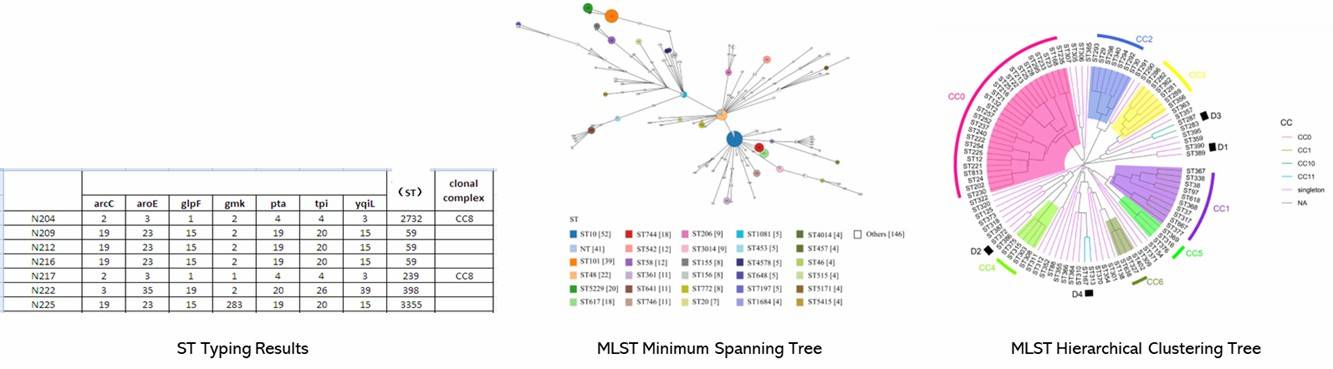

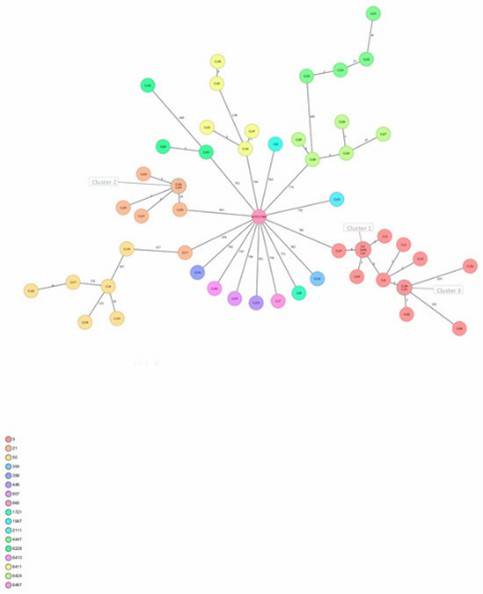 Figure 1. Minimum spanning tree of C. jejuni isolates generated.
Figure 1. Minimum spanning tree of C. jejuni isolates generated.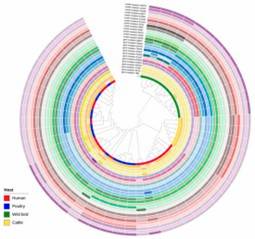 Figure 2. Interactive tree of life (iTOL) of 53 C. jejuni isolates based on the presence and absence of gene point mutations and sequence cluster classification.
Figure 2. Interactive tree of life (iTOL) of 53 C. jejuni isolates based on the presence and absence of gene point mutations and sequence cluster classification.