
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
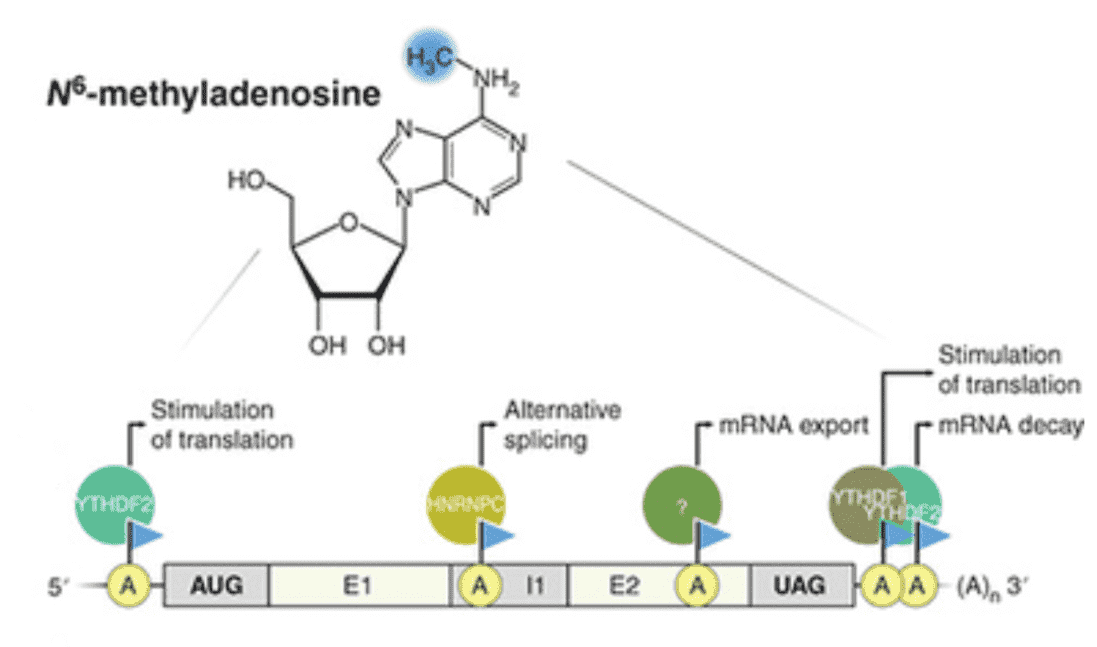
The most common DNA alteration in prokaryotes, but rare in eukaryotes, is DNA N6-methyladenine (6mA). Only specific unicellular eukaryotic organisms. Research has indicated the existence of 6mA from the newest advancements of highly sensitive identification techniques. Next-generation sequencing (NGS) offers another useful weapon to obtain essential genome-wide allocation details for these DNA adjustments in addition to advanced mass spectrometry that can efficiently evaluate the number of DNA modifications. For sequencing research, 6mA specific antibodies have been employed to identify and enhance 6mA-containing DNA fragments. By combining immunoprecipitation (IP) with NGS, 6mA can be assessed. But a quantitative template at single-base resolution with data on the alteration proportion is more beneficial. For the sequencing of methylated adenines, a bisulfite-treatment equivalent of a deamination strategy has yet to be improved. Although single-molecule real-time (SMRT) sequencing can differentiate 6mA from A and has been effectively implemented to small-genome prokaryotes, the significantly higher cost and reduced throughput have barred its broad usage in a much bigger genome eukaryotic lifeform.

We are dedicated to providing outstanding customer service and being reachable at all times.
Long-read sequencing has made a major contribution to the identification of the 6mA genome-wide allocation and can accomplish single-nucleotide resolution. Since the absolute 6mA abundance is considerably lower, adequate coverage greater than 100x is essential. SMRT sequencing enables the direct identification of altered nucleotides, such as 6mA, 5mC, and 5-hydroxymethylcytosine, in the DNA framework and does not significantly impair main DNA sequence determination. Using RS Modification and Motif Analysis, base adjustments (6mA) are defined.
From the current methods employed to identify adenosine methylation in RNA, 6mA DNA immunoprecipitation accompanied by deep sequencing (6mA DIP-Seq) has been established. We formed and incorporated this procedure for 6mA identification in DNA. In this procedure, with an antibody that identifies 6mA in genomic DNA, genomic DNA is extracted, fragmented and then DNA involving 6mA is brought down. DNA fragments that do not encompass 6mA are removed after subsequent washing and the 6mA comprising fragments are separated from the antibody to be further analyzed for subsequent analysis.

Our bioinformatics analysis bioinformatics analysis services are flexible to your specific projects.
| BIOINFORMATICS ANALYSIS | DETAILS |
|---|---|
| Genome Alignment | Integrating the raw sequence against the reference genome read from a FASTQ or unaligned BAM (uBAM) file. |
| CNA | By utilizing the depth of coverage strategy, approximate copy number changes (CNA) from aligned sequencing reads. |
| SV | Allow large structural variants to be identified, such as gene fusions and instability of microsatellites |
| Split-read alignment | Distinguishes gene fusions from the sequencing of genomic DNA |
| VCF | Variant classification outcomes are processed in one of the variant call mediums, enabling the processing of quantitative variant data, such as variant allele fraction, variant position coverage intensity, and genotype quality. |
| Queries across multiple genomic databases | To obtain tangible gene and variant nomenclature details |

Sampling kits: We provide a range of microbial sampling kits for clients, including MicroCollect™ oral sample microbial collection products and MicroCollect™ stool sample collection products.
References
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to info@cd-genomics.com for inquiries.
Please fill out the form below: ×


Follow us on: