- Home
- Solutions
- Epigenome Sequencing
- Metagenomics Sequencing
- DRUG-seq Service
Human leukocyte antigen typing (HLA typing) is a crucial process for determining the specificity of an individual's HLA genes. HLA, located on human chromosome 6, encodes glycoprotein molecules on the cell surface and plays a fundamental role in the immune system. It holds great significance in organ transplantation, disease correlation research, anthropological studies, and immunotherapy.
The HLA gene complex encompasses multiple gene loci, which are classified into three categories: class I, class II, and class III genes. Class I genes, including HLA-A, HLA-B, and HLA-C, are primarily expressed on the surface of nucleated cells. The molecules they encode present endogenous antigens to CD8⁺ T cells, thereby participating in the cellular immune response and playing a vital role in identifying and eliminating virus-infected cells and tumor cells. Class II genes, such as HLA-DR, HLA-DQ, and HLA-DP, are mainly expressed on the surface of antigen-presenting cells (e.g., macrophages, dendritic cells, and B cells). They are responsible for presenting exogenous antigens to CD4⁺ T cells, initiating the immune response, and are of key importance in regulating the intensity and type of immune response. Class III genes encode other molecules related to immunity, such as complement components, which are involved in inflammatory reactions and immune regulation.
Serological methods were used to detect lymphocyte surface antigens with known anti-HLA antibodies. Cytological method was used to observe the cell reaction typing by mixed lymphocyte culture. Molecular biological methods, such as PCR-SSP and PCR-SSOP, amplify and analyze HLA genes with high accuracy.
Serological method: Traditionally, the serological method relies on the combination of anti-HLA antibodies with known specificities and HLA antigens on the lymphocyte surface. It is detected through complement-dependent cytotoxicity tests (CDC) or microlymphocytotoxicity tests (MLCT). In the CDC test, lymphocytes to be detected are mixed with a series of known anti-HLA sera. If the antibodies in the sera bind to the corresponding HLA antigens on the lymphocyte surface, the lymphocytes will be damaged or killed in the presence of complement. The HLA type can then be determined by observing the death of the cells. MLCT is a more sensitive serological detection method capable of detecting weak antigen-antibody reactions. However, serological methods have certain limitations. They can only detect HLA antigens that have already been identified, and some rare or new alleles may not be accurately recognized. Additionally, the specificity and affinity of antibodies can vary, affecting the accuracy of the detection results.
PCR-based technology: Polymerase chain reaction (PCR)-related technologies are widely utilized in HLA typing. For example, sequence-specific primer PCR (PCR-SSP) employs primers designed for specific HLA allele sequences to amplify the sample under test. If an allele sequence complementary to the primer exists in the sample, a specific amplification product will be generated. The HLA type can be determined by detecting the presence or absence of the amplification product through gel electrophoresis. This method offers high specificity and relatively straightforward operation. Nevertheless, it requires the design of a large number of primers and can only detect known alleles. Sequence-specific oligonucleotide probe hybridization (PCR-SSOP) involves amplifying HLA gene fragments first and then hybridizing them with a series of labeled sequence-specific oligonucleotide probes. The HLA type is judged based on the hybridization signal. Its advantage lies in the ability to detect multiple alleles simultaneously, but the operation is complex, and the optimization of hybridization conditions significantly impacts the accuracy of the results.
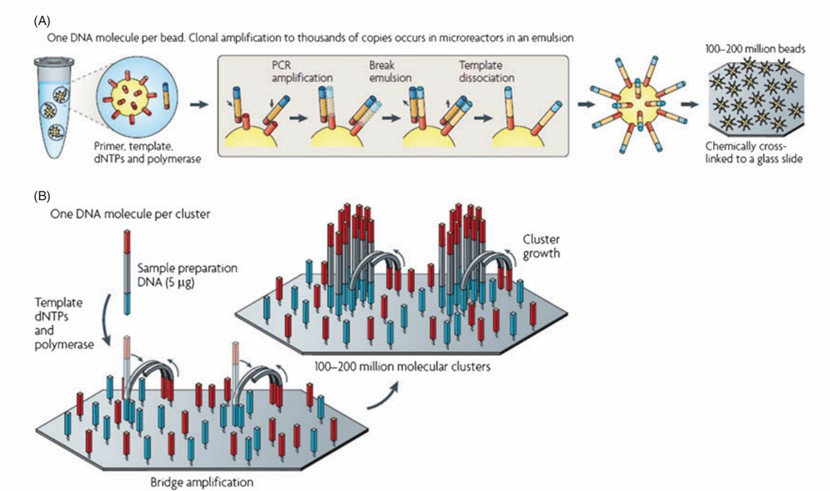 Preparation of HLA typing by clonal amplification by PCR (Erlich., 2012)
Preparation of HLA typing by clonal amplification by PCR (Erlich., 2012)
Method based on DNA sequencing: Direct sequencing of the HLA gene is an extremely accurate approach that can determine the nucleotide sequence of the HLA gene, enabling precise identification of alleles. For instance, the Sanger sequencing method can detect both known and unknown allele variations by sequencing the amplified products of HLA genes. Next-generation sequencing (NGS) technology has made it possible to perform high-throughput sequencing of multiple HLA loci in a single reaction, substantially enhancing the detection efficiency and resolution. It can comprehensively analyze the polymorphism of HLA genes. However, it comes with relatively high costs and more complex data analysis.
A study published in HLA Immune Response Genetics introduced a personalized HLA typing method. This method can identify new HLA alleles and tumor-specific HLA variants from whole exome sequencing (WES) data, presenting a more accurate and comprehensive solution for the application of HLA typing in clinical and research settings.
Research background
HLA full-length typing is of significant clinical and research value. However, due to the complexity and polymorphism of the HLA region, achieving accurate typing remains challenging. NGS data is commonly used for HLA typing, and the methods can be divided into HLA targeted sequencing and standard NGS sequencing (such as WES, whole genome sequencing (WGS), RNA-Seq), each with its own pros and cons. Most HLA typing tools based on NGS rely on database matching, which has limitations, particularly in detecting new alleles. In cancer, a comprehensive characterization of the HLA status is crucial for immunotherapy, but existing methods are insufficient.LA status is very important for immunotherapy, but the existing methods are insufficient.
Experimental method
Experimental results
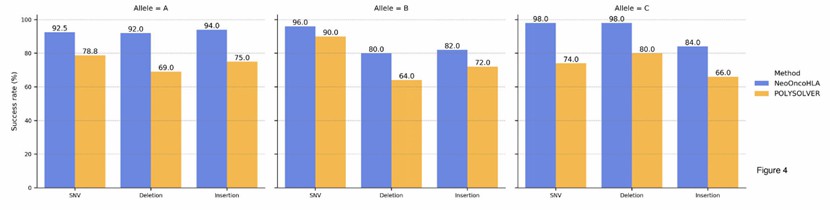 Somatic HLA variant simulation results (Anzar et al., 2022)
Somatic HLA variant simulation results (Anzar et al., 2022)
Discussion
The HLA typing method described in this paper is based on NGS data and combined with the comparison of known HLA allele databases. It can identify new alleles and tumor-specific variants and improving typing accuracy. Compared with other methods, it has advantages in the discovery of new alleles and the detection of tumor-specific variants. However, it is limited by the sequence similarity to known alleles and has mainly been tested on classical class I alleles. New methods may be required for the detection of highly divergent new alleles, and the germline variation detection steps can be further optimized. This method is beneficial for enriching the HLA library, enhancing the accuracy of HLA typing in clinical and research applications, and is of great significance for understanding the mechanism of tumor immune escape and developing cancer immunotherapy strategies.
Take the Next Step: Explore Related Services
Discover More: Recommended Reads
A paper published in Bioinformatics introduced a novel HLA genotyping algorithm called OptiType. This algorithm can accurately predict HLA genotypes from NGS data without specific enrichment of the HLA region and performs well across multiple datasets, providing a fast, accurate, and economical method for HLA genotyping. It is expected to play an important role in clinical applications.
Research background
The HLA gene cluster is of utmost importance in adaptive immunity and is closely related to vaccinology, regenerative medicine, transplantation medicine, autoimmune diseases, and other fields. HLA alleles exhibit high polymorphism and sequence similarity. Traditional HLA typing methods have limitations, such as being labor-intensive, time-consuming, and yielding unclear results. Although there are HLA typing methods based on NGS, issues such as cumbersome preparation, long processing times, or insufficient accuracy still exist. In particular, the computational methods for determining HLA genotypes from conventional sequencing data are not accurate enough.
Experimental method
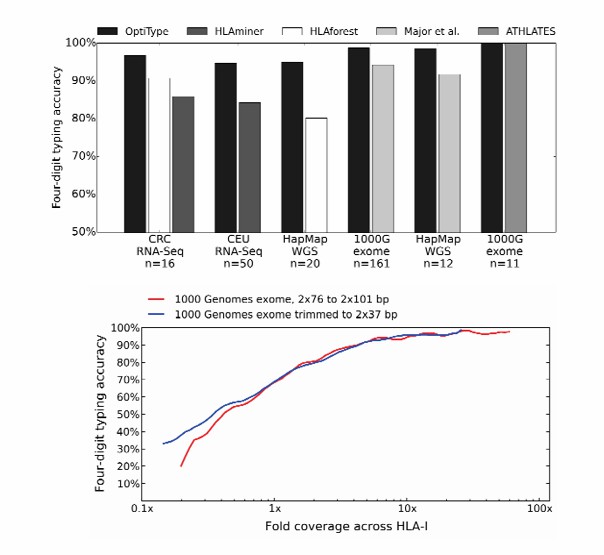 Performance comparison of HLA typing algorithms (Szolek et al., 2014)
Performance comparison of HLA typing algorithms (Szolek et al., 2014)
Experimental result
Discussion
OptiType is a fast and accurate HLA typing method based on NGS data. It can perform automatic typing with four-digit resolution and performs well on different types of sequencing data, making it suitable for NGS data from various sources. It is superior to previous computational methods. Coverage depth above a certain level has minimal impact on its performance. Short read lengths increase mapping ambiguity but do not affect the method's performance. Incorrect predictions are mainly caused by issues such as uncovered sequences, failed zygosity detection, and ambiguous typing of small loci. Ensuring that each allele has an equal opportunity to be identified is crucial. Using phylogenetic methods to reconstruct intron sequences can help improve performance. With the increase in the number of fully sequenced HLA alleles, the reference sequence can be expanded, which is expected to enhance the prediction accuracy. OptiType provides an alternative to common HLA genotyping methods, although it can only predict known alleles.
A study on HLA typing focused on the differences in HLA typing among hospitalized children with sickle cell disease (SCD). The details are as follows:
Research background
Treatment: Hematopoietic stem cell transplantation (HSCT) is an effective treatment for SCD, and HLA typing is the critical first step in HSCT.
Current situation: The proportion of SCD patients who have completed HLA typing is unclear. Socio-economic factors may influence the HLA typing of SCD families and their access to HSCT, resulting in a medical disparity.
Research method
Research results
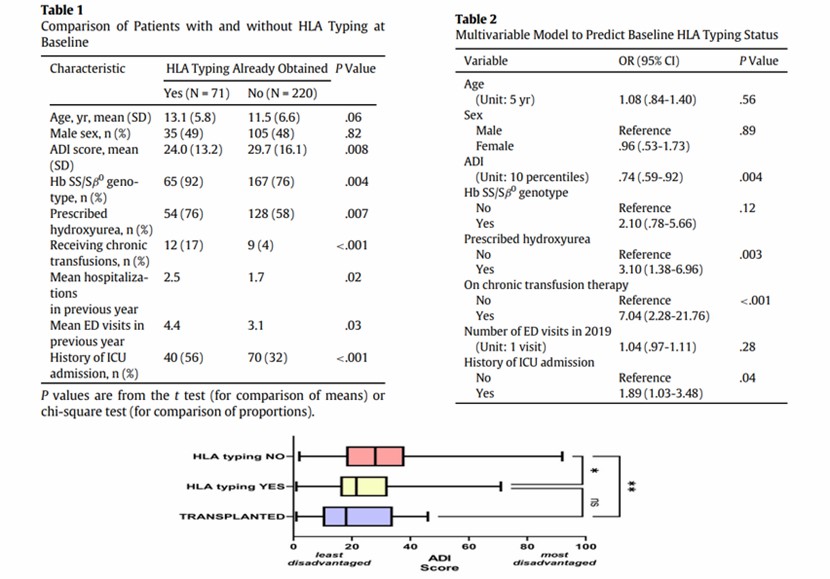 Comparison of ADI in hospitalized patients and patients who underwent HSCT for SCD (Takor et al., 2024)
Comparison of ADI in hospitalized patients and patients who underwent HSCT for SCD (Takor et al., 2024)
Discussion
Classification status: Most hospitalized SCD patients (>75%) had not undergone HLA typing, and patients with more severe diseases were more likely to be typed, leading to a medical gap related to socio-economic status.
Acceptance: Most (69%) families who had not had HLA typing at baseline expressed interest in testing, suggesting that doctors may not have fully informed patients about HLA typing and HSCT options.
Limitations of the study: The study only involved one institution, and the results may not be generalizable. In the future, it is necessary to ensure that all SCD patients can receive equal treatment.
With the continuous advancement of technology, HLA typing is expected to become more accurate, efficient, and widely used in the future. New molecular biology techniques and data analysis algorithms will continue to emerge, further enhancing the resolution and accuracy of typing, reducing costs, and shortening detection times. Additionally, the sharing and collaborative research of HLA typing data worldwide will continue to strengthen, contributing to a deeper understanding of the diversity and function of human HLA genes and promoting the development of related fields.
References
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.



