
 Sample Submission Guidelines
Sample Submission Guidelines
Bioinformatics Workflow for Whole Genome Sequencing
Overview of WGS
Whole Genome Sequencing (WGS) refers to the high-throughput sequencing of the entire genome, allowing for the analysis of variations between different individuals, as well as the annotation of single nucleotide polymorphisms (SNPs) and genomic structural features. WGS, owing to its comprehensive nature, provides a wealth of information beyond what is attainable through exome sequencing or targeted sequencing, thereby offering unique advantages.
Furthermore, advancements in sequencing technologies in recent years coupled with the continual reduction in sequencing costs have made WGS increasingly accessible. Its superiority in identifying single nucleotide polymorphisms (SNPs), insertions, and deletions (Indels) further underscores its significance, rendering WGS an increasingly favored option in both clinical and fundamental research endeavors.
WGS has the capacity to greatly enhance genomic knowledge and understand mysteries of life by utilizing the most advanced genetic sequencing technologies. WGS can be used for variant calling, genome annotation, phylogenetic analysis, reference genome construction, and more. WGS tries to cover the whole genome, but actually covers 95% of the genome with technical difficulties in sequencing regions such as centromeres and telomeres. Another challenge for WGS is data management. As larger datasets become more accessible and affordable, computational analysis will be the rate-limiting factor rather than sequencing technology. Here we will discuss the bioinformatics workflow for detection of genetic variations in WGS to help you get through it.
Service you may intersted in
Bioinformatics Workflow for WGS
The bioinformatics workflow for WGS is similar to that for whole exome sequencing. You can view our article Bioinformatics Workflow for Whole Exome Sequencing. The bioinformatics workflow for WGS falls into the following steps: (1) raw read quality control; (2) data preprocessing; (3) alignment; (4) variant calling; (5) genome assembly; (6) genome annotation; (7) other advanced analyses based on your research interest such as phylogenetic analysis.
 Figure 1. Bioinformatics workflow of whole genome sequencing.
Figure 1. Bioinformatics workflow of whole genome sequencing.
Raw read QC and preprocessing
The relevance of quality control lies in its application to next-generation sequencing (NGS), primarily represented by Illumina, which principally employs the technique of sequencing by synthesis. The fabrication of nucleotides relies on chemical reactions, allowing for a ceaseless synthesis and extension of the nucleotide chain from the 5' end towards the 3' end.
However, throughout this synthesis process, the efficacy of DNA polymerase invariably wanes concurrent with the growth of the synthesis chain, and its specificity commences to decline. This inevitably manifests a predicament — as we progress further, the replication error rate of the nucleotides intensifies. The quality of sequencing data inherently influences our downstream analysis signalling the significance of stringent quality controls.
Data directly obtained from the sequencer involves all nucleotides, irrespective of their sequencing quality. They may also contain errors, and possibly includes experimental inaccuracies. Among quality control steps, raw sequencing data is input into quality control software, where low-quality or unsequenced, and mis-sequenced nucleotides are eliminated. These steps yield QC filtered low-quality read data (clean data).
Clean data is accordingly considered devoid of sequencing errors. Suppose our raw data comprises reads (10,000 units); after quality control is applied, this quantity will transform into clean data (8,500 units).
The raw files (fastq) need to be eliminated from poor-quality reads/sequences and technical sequences such as adapter sequences. This process is important for accurate and reliable variation detection. FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastq) is a powerful tool for raw read QC that generates statistics data results, including basic statistics, sequence quality, quality scores, sequence content, GC content, sequence length distribution, overrepresented sequences, sequence duplication level plots, adapter content, and k-mer content. Tools like Fastx_trimmer and cutadapt can be used for read trimming.
Alignment
Alignment refers to the positioning of short sequenced fragments against a known reference genome sequence to determine the location and potential variations of each fragment within the genome.
Alignments can help pinpoint the precise location of sequencing fragments on the reference genome, thereby revealing specific regions and structures within the genome. By alignment, differences between the sequenced sample and reference genome help identify Single Nucleotide Polymorphisms (SNPs), insertions and deletions mutations, and so forth. Alignment is also a critical step in gene annotation; it aids in determining the regions of genes such as exons, introns, promoters, exons, and UTRs.
A reference genome needs to be determined. Mash enables us to compare the sequencing reads generated against the reference set from NCBI RefSeq genomes (https://www.ncbi.nlm.nih.gov/refseq) to determine genetic distance and relatedness. The next step is to map the quality-controlled reads to the reference genome. Burrows-Wheeler Aligner (BWA) and Bowtie2 are two popular short read alignment algorithms. The output of BWA and Bowtie2 is the standard sequence alignment/map format known as SAM, which facilitates the following steps. Alternatively, BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) is widely used for local alignment.
Table 1. The common computational programs for read alignment.
| Program | Source type | Website |
|---|---|---|
| Bowtie2 | Open source | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| SEAL | Open source | http://compbio.case.edu/seal/ |
| SOAP3 | Open source | http://www.cs.hku.hk/2bwt-tools/soap3/; http://soap.genomics.org.cn/soap3.html |
| BWA, BWA-SW | Open source | http://bio-bwa.sourceforge.net/ |
| Novoalign | Commercially available | http://www.novocra.com/ |
| SHRiMP/SHRiMP2 | Open source | http://compbio.cs.toronto.edu/shrimp/ |
| MAQ | Open source | http://maq.sourceforget.net/ |
| Stampy | Open source | http://www.well.ox.ac.uk/project-stampy/ |
| ELAND | Commercially available | http://www.illumina.com/ |
| SARUMAN | Open source | http://www.cebitec.uni-bielefeld.de/brf/saruman/saruman.html |
Variant calling
Once reads are aligned to the reference genome, variants can be identified by comparing the sample genome to the reference genome. Detected variants may be associated with disease, or simply be non-functional genomic noise. Variant call format (VCF) is the standard format for storing sequence variations, including SNPs (single nucleotide polymorphisms), indels, structural variants, and annotations. Variant calling can be complicated due to high rate of false positive and false negative identifications of SNVs and indels. The software packages in Table 2 are useful for improving variant calling.
Table 2. The software packages for variant calling.
| Software packages | Descriptions | Website |
|---|---|---|
| GATK |
|
http://software.broadinstitute.org/gatk/ |
| SOAPsnp |
|
http://soap.genomics.org.cn/ |
| VarScan/VarScan2 |
|
http://genome.wustl.edu/tools/cancer-genomics |
| ALTAS 2 |
|
http://www.genboree.org/ |
In whole-genome sequencing (WGS) analysis, variant detection heavily depends on the quality score of sequencing bases because this score is an integral (sometimes the sole) standard for measuring the accuracy of our sequenced bases. The Base Quality Score Recalibration (BQSR) primarily constructs error models for sequencing bases through machine learning and makes appropriate adjustments to these base quality scores.
Lastly, quality control and filtering of the variant results are necessary. The purpose of quality control is to discard false-positive results to the maximum extent possible while retaining as much accurate data as possible. The preferred quality control scheme is the GATK VQSR (Variant Quality Score Recalibration), which uses machine learning to train a model (Gaussian mixture model) using various data features, allowing for quality control of variant data.
Genome assembly
De novo assembly is the process to align overlapping reads to form longer contigs (larger contiguous sequences) and order the contigs into scaffolds (a framework of the sequenced genome). If there is a reference genome from a related species, the common method is to first generate contigs de novo and then align them to the reference genome for scaffold assembly. An alternative approach is the "Align-Layout-Consensus" algorithm. This method first aligns reads to a closely related reference genome, and then builds contigs and scaffolds de novo.
Table 3. The common assemblers for diverse sequencing platforms.
| Sequencing platform | Tools for genome assembly |
|---|---|
| Illumina | Velvet (https://www.ebi.ac.uk/~zerbino/velvet/) SPAdes (http://bioinf.Spbau.Ru/spades) |
| Ion Torrent | MIRA (http://www.Chevreux.Org/projects_mira.html) |
| Roche 454 | Newbler (http://454.com/contact-us/software-request.asp) |
| PacBio SMRT | SPAdes, HGAP, and the Celera-MHAP assembler |
Users can assess the quality of draft genome assemblies or compare assemblies generated by different methods. There are a variety of metrics that reflect the quality of the assembly. Only contiguous near-complete (approximately 90%) assembly interrupted by small gaps will yield successful genome annotation.
- Genome size. Both C-value and k-mer frequency-based approaches can infer genome size.
- Assembly contiguity. N50 statistic can be used to evaluate assembly contiguity, which describes a kind of median of assembled sequence lengths.
- Accuracy. Transcriptome data present an important resource for validating sequence accuracy and correcting scaffolds. Comparative genomic approaches can also provide guidance in detecting mis-assemblies and chimeric contigs.
Genome annotation
To fully understand the genome sequence, it needs to be annotated with biologically relevant information such as gene ontology (GO) terms, KEGG pathways, and epigenetic modifications. The annotation involves two phases:
(1) Computational phase. A computational phase includes repeat masking, prediction of coding sequence (CDS), and prediction of gene models.
- Repeat masking. Since repeats are poorly conserved across species, it is recommended to create a species-specific repeat library by utilizing tools such as RepeatModeler, RepeatExplorer.
- Prediction of CDS. Predict CDS using ab initio algorithms.
- Prediction of gene models. Protein alignment, syntenic protein lift-overs from other species, EST, and RNA-seq data can provide a valuable resource for predicting gene models.
(2) Annotation phase. All the evidence mentioned above (ab initio prediction, as well as protein-, EST-, and RNA-alignments) is then synthesized into a gene annotation. Additionally, automated annotation tools such as MAKER and PASA are available to integrate and weigh the evidence. WebApollo can be used to edit the annotation through the visual interface if anything is wrong with the gene annotations.
Once the genome annotation is assessed by visual inspection, you can publish the draft genome sequences and annotation. In order to allow others to improve the genome assembly and annotation, all raw data should be uploaded. The available databases for uploading genome include ENSEMBL and NCBI.
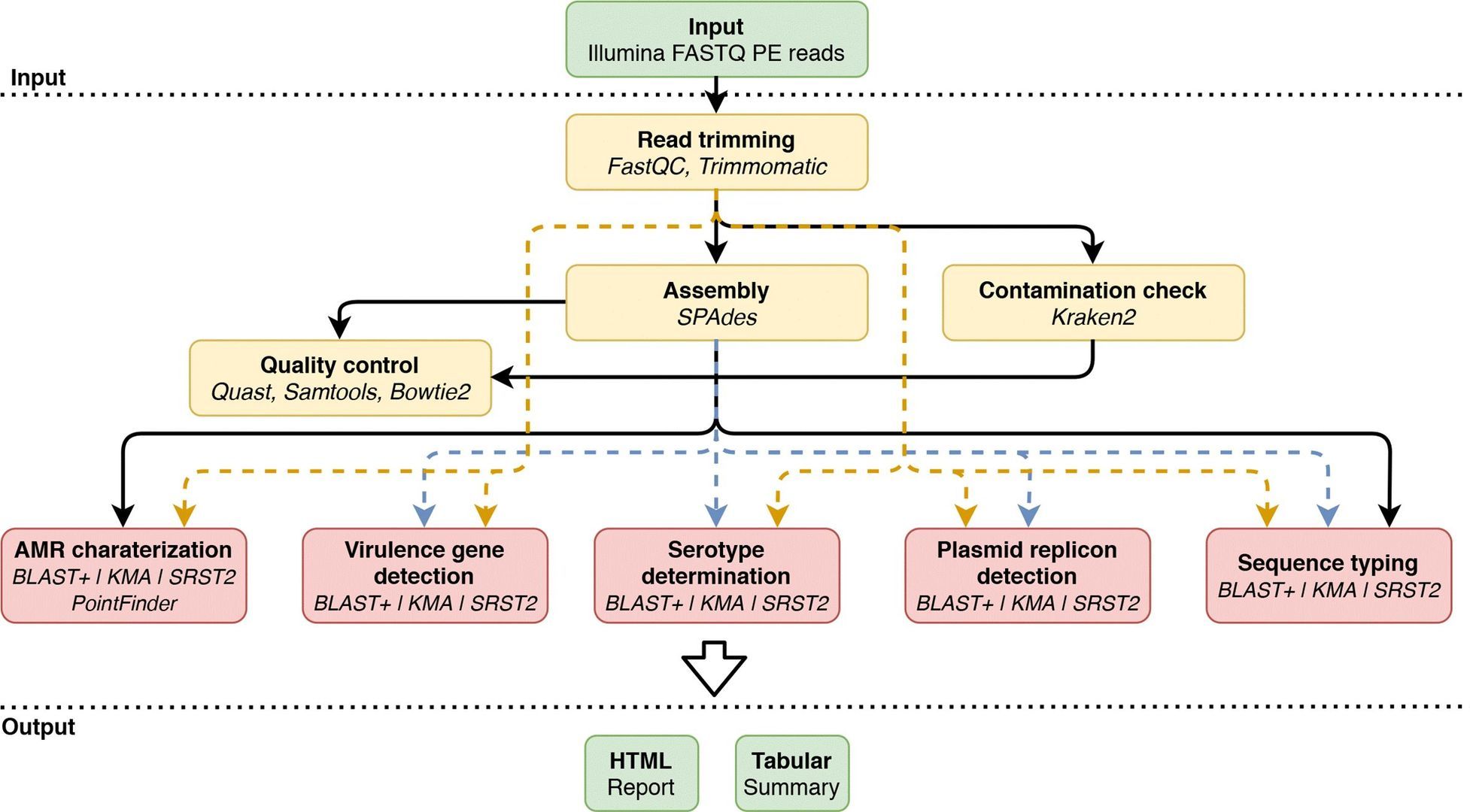 Figure 2. Overview of the bioinformatics workflow. (Bogaerts et al., 2021)
Figure 2. Overview of the bioinformatics workflow. (Bogaerts et al., 2021)
Other advanced analyses
Additional aspects of whole-genome sequencing results can be analyzed, such as utilizing tools like Staramr for genomic genotype identification. This includes Multilocus Sequence Typing (MLST) and Core Genome Multilocus Sequence Typing (cgMLST). Furthermore, databases like ResFinder are employed for the detection of antimicrobial resistance genes.
Tools such as PlasmidFinder are utilized to detect plasmid replicons, thereby analyzing the type and distribution of plasmids. Using ABRicate, in conjunction with virulence factor databases (such as VFDB), facilitates the detection of genes associated with bacterial virulence.
Softwares like Roary are used to construct the core genome and pan-genome, while tools such as IQ-TREE assist in the development of phylogenetic trees, helping to analyze the evolutionary relationships between strains. To visualize phylogenetic trees and metadata, tools like iTOL are implemented, which generate reports that are easily comprehensible.
If you are interested in our genomics services, please visit our website: www.cd-genomics.com for more information. We can provide a full package of genomics sequencing, including whole genome sequencing, whole exome sequencing, targeted region sequencing, mitochondrial DNA (mtDNA) sequencing, and complete plasmid DNA sequencing.
References:
- Dolled-Filhart M P, Lee M, Ou-yang C, et al. Computational and bioinformatics frameworks for next-generation whole exome and genome sequencing. The Scientific World Journal, 2013, 2013.
- Ekblom R, Wolf J B W. A field guide to whole‐genome sequencing, assembly and annotation. Evolutionary applications, 2014, 7(9): 1026-1042.
- Kwong J C, McCallum N, Sintchenko V, et al. Whole genome sequencing in clinical and public health microbiology. Pathology, 2015, 47(3): 199-210.
- Meena N, Mathur P, Medicherla K M, et al. A Bioinformatics Pipeline for Whole Exome Sequencing: Overview of the Processing and Steps from Raw Data to Downstream Analysis. bioRxiv, 2017: 201145.
- Oakeson K F, Wagner J M, Mendenhall M, et al. Bioinformatic analyses of whole-genome sequence data in a public health laboratory. Emerging infectious diseases, 2017, 23(9): 1441.
- Atxaerandio-Landa A, Arrieta-Gisasola A, Laorden L, et al. A Practical Bioinformatics Workflow for Routine Analysis of Bacterial WGS Data. Microorganisms. 2022 Nov 29;10(12):2364.
- Bogaerts B, Nouws S, Verhaegen B, et al. Validation strategy of a bioinformatics whole genome sequencing workflow for Shiga toxin-producing Escherichia coli using a reference collection extensively characterized with conventional methods. Microbial Genomics, 2021, 7(3): 000531.
- Bogaerts B, Delcourt T, Soetaert K, et al. A Bioinformatics Whole-Genome Sequencing Workflow for Clinical Mycobacterium tuberculosis Complex Isolate Analysis, Validated Using a Reference Collection Extensively Characterized with Conventional Methods and In Silico Approaches. Journal of Clinical Microbiology, 2021, 59(6): 10.1128/jcm. 00202-21.


