
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics is providing PacBio Single Molecular Real-Time (SMRT) sequencing to increase your research method for bacterial whole genome sequencing. A comprehensive view of the bacterial genome, including genes, regulatory regions, IS elements, phage integration sites, and base modifications is vital to understanding key traits such as antibiotic resistance, virulence, and metabolism.
Introduction of Bacterial Whole Genome de novo Sequencing
The process of de novo bacterial genome sequencing, also known as whole-genome de novo sequencing, involves sequencing a bacterial genome without the reliance on any reference sequence information. This is accomplished through advanced bioinformatics techniques that facilitate the assembly and alignment of the sequencing data, ultimately yielding a comprehensive map of the organism's genome. This map is essential for subsequent structural and functional analyses.
Third-generation sequencing technologies, such as Single Molecule Real-Time (SMRT) sequencing, are particularly adept at sequencing individual molecules and generating significantly longer reads, often exceeding 15,000 base pairs and, in some cases, surpassing 100,000 base pairs. These long-read capabilities are instrumental in overcoming the challenges posed by repetitive sequences and regions with high GC content, which are common difficulties in genome assembly. SMRT sequencing can thus generate complete bacterial genome assemblies devoid of gaps and undefined bases (Ns).
The merits of SMRT sequencing are particularly pronounced when compared to short-read platforms such as Illumina HiSeq, which utilizes sequencing-by-synthesis technology. Microbial whole genome sequencing by Illumina HiSeq platforms utilizes sequencing by synthesis technology. Illumina platforms, although highly accurate, are constrained by their shorter read lengths, typically ranging from 50 to 300 base pairs. Furthermore, the required PCR amplification of multiple DNA templates prior to sequencing introduces potential base-composition biases, potentially skewing the GC content of the resulting sequences.
In contrast, the long reads produced by SMRT sequencing facilitate the assembly of bacterial genomes into single contigs, effectively addressing issues stemming from abnormal GC content and high duplication. Among various assembly methods, the PacBio assembly has demonstrated superior recovery of core genes, virulence factors, and housekeeping genes, as verified by whole-genome multilocus sequence typing. This comprehensive assembly capability accelerates research into the genetic structure and functional aspects of bacterial genomes, providing a robust foundation for exploring genetic and pathogenic properties.
Advantages of Bacterial Whole Genome de novo Sequencing
- Detailed information on genetic structure and function of species.
- Genome assembly minimally impacted by GC content and repetitive sequences; better integrity.
- Identification of strain-specific variations.
- Generation of accurate reference sequences.
Applications of Bacterial Whole Genome de novo Sequencing
- Pathogenic Gene Mining: Identifying pathogenic genes associated with human health, livestock safety, plant protection, forestry and agricultural security, and biological weed control.
- Industrial Bacteria Gene and Mechanism Analysis: Characterizing functional genes and high-yield/environmental adaptation mechanisms in new or dominant industrial bacterial strains related to yogurt fermentation, energy utilization, and pharmaceutical production.
- Evolutionary and Medicinal Mechanism Exploration: Investigating species evolutionary mechanisms at the whole genome or mitochondrial and chloroplast levels, the extreme environment adaptation and evolutionary theories of archaea, and the nutritional and medicinal mechanisms of edible fungi.
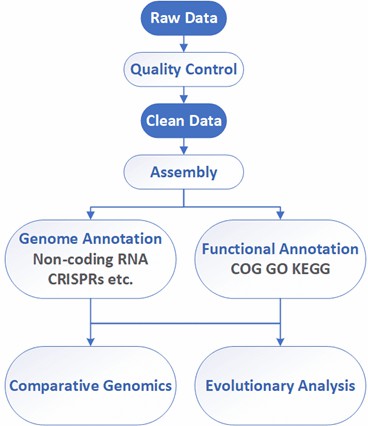
Bacterial Whole Genome de novo Sequencing Workflow
Our highly experienced expert team executes quality management by following every procedure to ensure comprehensive and accurate results. The general workflow for bacterial whole genome sequencing is outlined below.

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Bacterial Whole Genome de novo Sequencing for your writing (customization)
At CD Genomics, we are using the long-read PacBio Sequel platform to support researchers all over the word with bacterial de novo whole genome sequencing needs. As one of the most famous industry leaders who are skilled in sequencing, CD Genomics not only concentrates on developing cutting-edge technology but also be willing to share our state-of-the-art platforms and sufficient expertise with our clients to promote their brilliant studies. It is our guarantee to provide customers with the best services and finest results at CD Genomics.
Partial results are shown below:
1. What indicators can be used to evaluate bacterial genome assembly?
The common indicators for the quality of genome assembly include scaffold N50, N%, scaffold numbers, and the total number of base pairs.
2. How to achieve zero gap?
Currently, the complete sequence map of more than 90% bacterial strains can be constructed by making use of a combination of Illumina HiSeq and PacBio SMRT systems. Pacbio RS II system can achieve complete genome assembly even in the regions of high or low GC content, as well as repetitive sequences. The complete sequence map of the rest 10% bacterial strains can be achieved with Sanger sequencing data. CD Genomics has completed hundreds of bacterial genome assembly cases without gap.
3. Is it feasible to complete a bacterial genome using only third-generation single-molecule sequencing platforms?
No, it is not feasible. Small plasmid fragments (approximately 20 kb) may be lost during the library construction process. Additionally, certain regions of the chromosome may not be sequenced due to sampling probability issues or sample degradation.
4. How can we ensure the accuracy of the assembly given the low single-base accuracy of third-generation single-molecule sequencing platforms?
The single-base accuracy of third-generation single-molecule sequencing data ranges between 87% and 92%. To ensure the accuracy of the assembly, we can employ the following three-step process:
- Prior to assembly, correct the sequencing data by leveraging the overlap between third-generation single-molecule sequencing sequences.
- Post-assembly, use third-generation single-molecule sequencing data to correct the assembled sequences.
- After the second correction, use high-quality second-generation high-throughput sequencing data for further correction of the assembled results.
By applying this three-step correction process, the final assembly accuracy can exceed 99.99%.
PacBio But Not Illumina Technology Can Achieve Fast, Accurate and Complete Closure of the High GC, Complex Burkholderia pseudomallei Two-Chromosome Genome
Journal: Frontiers in microbiology
Impact factor: 4.235
Published: 02 August 2017
Background
Bacterial genome sequencing has evolved from labor-intensive Sanger sequencing to high-throughput Illumina platforms, offering accuracy but shorter read lengths. PacBio RS introduced longer reads with SMRT technology, albeit with higher error rates initially. Recent improvements in PacBio's chemistry and software have enhanced accuracy and read lengths. Comparing PacBio RS II and Illumina HiSeq 1500 in sequencing Burkholderia pseudomallei highlights their capabilities and challenges in studying complex bacterial genomes.
Materials & Methods
Sample Preparation
- B. pseudomallei
- DNA extraction
Sequencing
- SMRTbell library preparation
- PacBio sequencing
- Illumina Library Preparation
- Next generation sequencing
- Genome assembly
- Genome annotation
- Mapping to reference genome
- Circular genome visualization
Results
Sequence data from PacBio and Illumina were used for hybrid assembly of the B. pseudomallei genome, resulting in 74 contigs. Two large contigs representing chromosomes I and II were assembled manually using Geneious R8. Annotation revealed 7,014 protein-coding sequences and 71 RNAs, with a significant portion categorized into subsystems such as Amino Acids, Carbohydrates, and Membrane Transport. Many protein-coding sequences remain unclassified, including a large number annotated as hypothetical proteins.
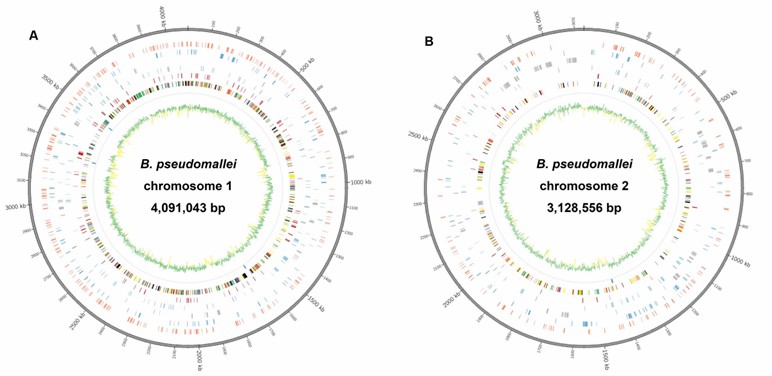 FIGURE 1. Circular representation of Burkholderia pseudomallei reference genome obtained by hybrid assembly.
FIGURE 1. Circular representation of Burkholderia pseudomallei reference genome obtained by hybrid assembly.
The PacBio assembly of B. pseudomallei strain 14M0960418 yielded two large scaffolds totaling 7,222,235 bp, with 7,014 protein-coding sequences, 71 RNAs, and 59 tRNA-encoding genes. The assembly had an average read length >8 kb, coverage depth of 143×, and a G+C content of 68.2%, and was syntenic with the reference genome.
TABLE 1. Genome characteristics for PacBio and Illumina platforms.
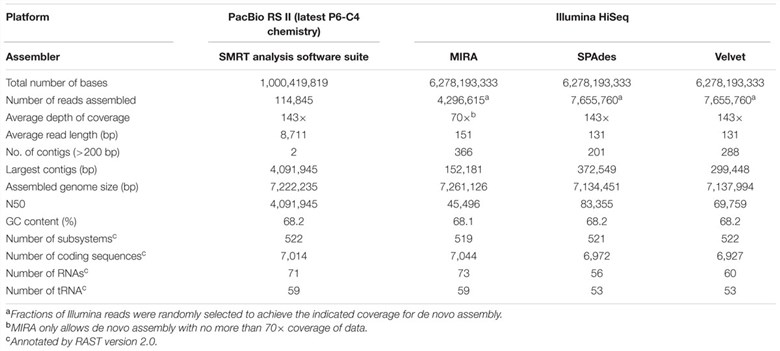
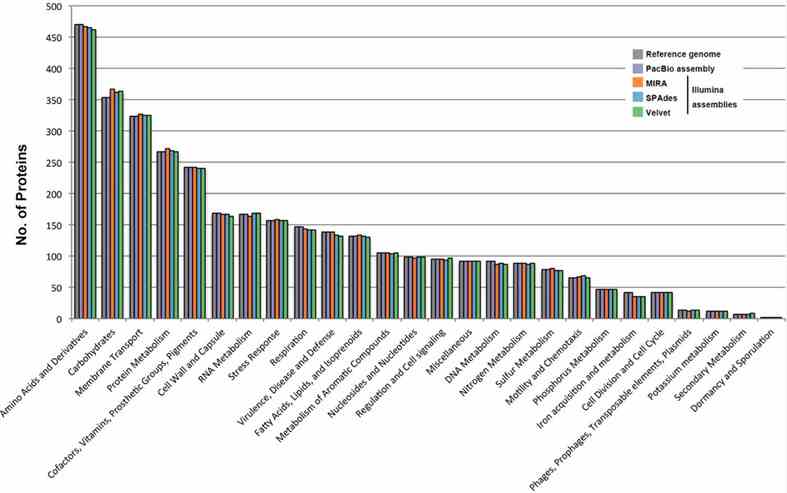 FIGURE 2. Comparison of genome sequence data of the B. pseudomallei strain from different assemblies.
FIGURE 2. Comparison of genome sequence data of the B. pseudomallei strain from different assemblies.
This study compared de novo assemblies from PacBio RS II and Illumina HiSeq data for B. pseudomallei. PacBio yielded two large, gapless contigs (4,091,945 and 3,130,290 bp), showing >99.9% identity with the reference genome and accurate genome organization. In contrast, Illumina assemblies were fragmented (MIRA: 366; SPAdes: 201; Velvet: 288 contigs), with varied nucleotide identities (92.2–100%). Illumina contigs also showed mis-assemblies and uncovered regions, contrasting with the cohesive PacBio assembly. Ribosomal operon prediction confirmed PacBio's accuracy, contrasting with partial detections in Illumina assemblies.
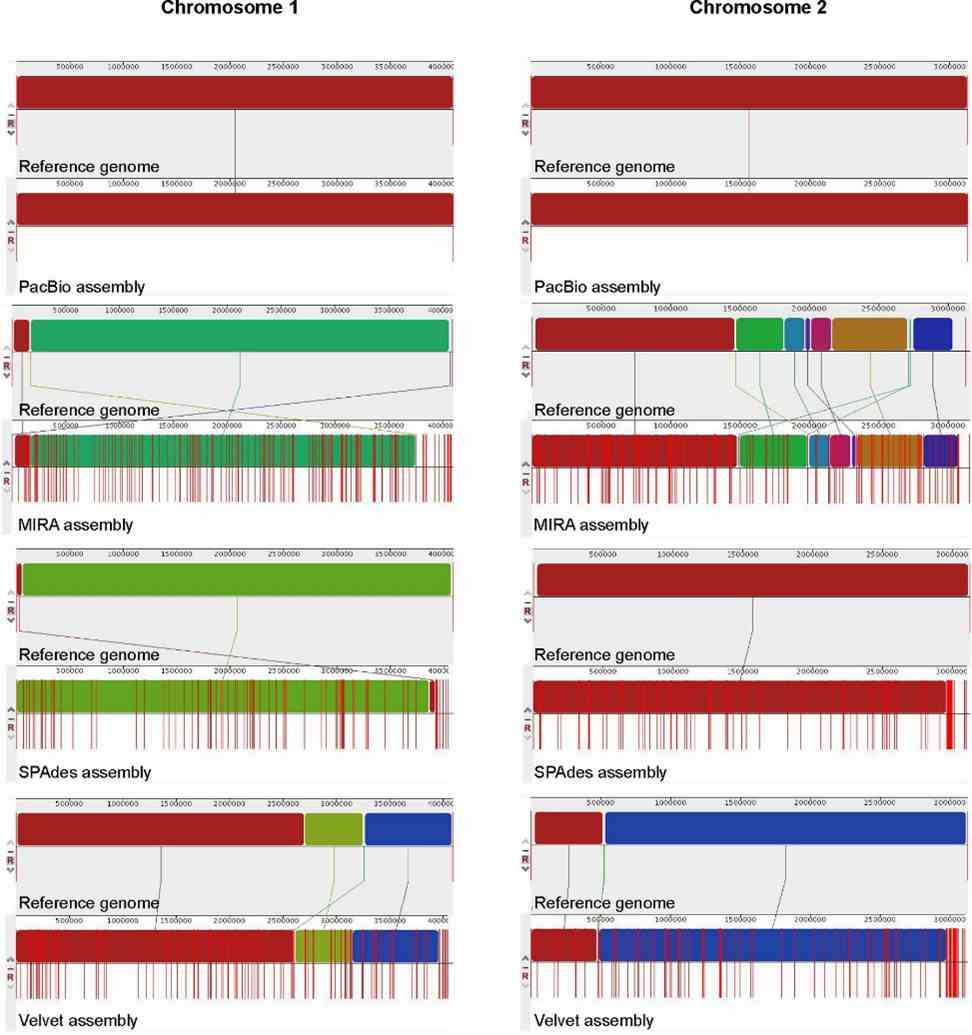 FIGURE 3. Whole genome alignment of B. pseudomallei genomes.
FIGURE 3. Whole genome alignment of B. pseudomallei genomes.
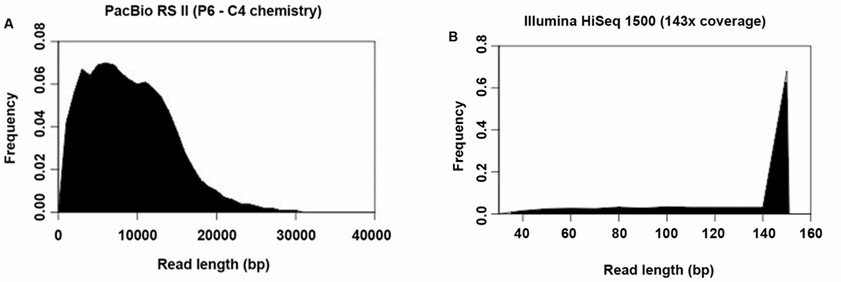 FIGURE 4. Histogram showing the read length distribution generated by (A) PacBio RS II and (B) Illumina HiSeq 1500.
FIGURE 4. Histogram showing the read length distribution generated by (A) PacBio RS II and (B) Illumina HiSeq 1500.
Conclusion
PacBio RS II with P6-C4 chemistry fully sequenced the B. pseudomallei genome accurately, revealing its two-chromosome structure and four ribosomal operons. In contrast, Illumina HiSeq 1500 produced fragmented contigs and mis-assemblies, particularly in complex and repetitive regions. PacBio's long reads (10,000-15,000 bases) ensured complete genome coverage, showcasing its cost-effectiveness and suitability for resolving challenging bacterial genomes like B. pseudomallei.
Reference
- Teng JL, Yeung ML, Chan E, et al. PacBio but not Illumina technology can achieve fast, accurate and complete closure of the high GC, complex Burkholderia pseudomallei two-chromosome genome. Frontiers in microbiology. 2017, 8:1448.
Here are some publications that have been successfully published using our services or other related services:
Identification of diverse integron and plasmid structures carrying a novel carbapenemase among Pseudomonas species
Journal: Front. Microbiol.
Year: 2019
Production of a Bacteriocin Like Protein PEG 446 from Clostridium tyrobutyricum NRRL B-67062
Journal: Probiotics and Antimicrobial Proteins
Year: 2024
Untangling the Role of Pathobionts from Bacteroides Species in Inflammatory Bowel Diseases
Journal: bioRxiv
Year: 2023
A chromosome-level genome resource for studying virulence mechanisms and evolution of the coffee rust pathogen Hemileia vastatrix
Journal: bioRxiv
Year: 2022
Streptomyces buecherae sp. nov., an actinomycete isolated from multiple bat species
Journal: Antonie Van Leeuwenhoek
Year: 2020
See more articles published by our clients.


