Introduction to Sex Chromosome Sequencing and Assembly
Sex chromosomes have been a fascinating research hotspot in evolutionary and developmental biology due to their specific and diverse evolutionary trajectories, including X/Y, Z/W and U/V sex chromosomes. However, sex chromosomes have been the more difficult and less effective regions for plant and animal genome assembly and annotation. With rapid improvements in genome sequencing, assembly and scaffolding technologies, it is now feasible to construct sex chromosome typing assemblies.
The sex chromosomes, comprising X and Y chromosomes, are pivotal determinants of biological sex, each playing distinct roles across various species. Sequencing and assembling sex chromosomes represent a complex yet indispensable endeavor, involving multiple procedural steps. Initially, sample collection and DNA extraction are imperative, followed by sequencing the extracted DNA using modern sequencing platforms such as Illumina, PacBio SMRT, and Oxford Nanopore. These sequencing technologies yield extensive DNA sequence fragments, laying the groundwork for subsequent analyses. Post-sequencing, meticulous processing and purification of generated sequence data are conducted to eliminate low-quality sequences and contamination. Subsequently, a combination of paired-end or long-read sequencing data is employed to enhance sequencing accuracy and quality. Through these sequencing efforts, identification and segregation of X and Y chromosome sequences are achieved, serving as the foundation for sex chromosome assembly and further comprehensive analysis.
Services you may interested in
What are the Strategies for Sex Chromosome Assembly
The analysis of sex chromosomes in different species is a crucial aspect of genomic research aimed at understanding sex determination mechanisms and genetic diversity. There are multiple strategies utilized in this process, each with its unique advantages and challenges.
The first approach involves individually sequencing the sex chromosomes of the species through targeted enrichment techniques, such as the micro-nuclear carrier method or single-cell sequencing. This method isolates the sex chromosomes from other chromosomes and conducts separate sequencing, thus enhancing the coverage and depth of the sex chromosomes. Consequently, this leads to more comprehensive and accurate assembly of the sex chromosomes.
The second strategy entails specifying sex chromosomes by increasing sequencing depth within the entire genome sequencing project. This augmentation ensures coverage of all regions of the sex chromosomes. By amplifying sequencing depth, the coverage and reliability of sex chromosomes are improved, thereby facilitating better assembly and analysis.
The final approach combines de novo sequencing with resequencing methods. Initially, de novo sequencing is employed to obtain preliminary assembly results of the sex chromosomes. Subsequently, resequencing technologies, such as long-read sequencing, are utilized to fill sequence gaps and resolve complex repetitive regions in the assembly. This integrated strategy enhances the continuity and accuracy of sex chromosome assembly, particularly in addressing complex repetitive sequences.
Y Chromosome Assembly in Gorillas and Humans
Mammalian Y chromosome sequences are critical for studying sex determination, but the Y chromosome is rich in repetitive and palindromic sequences and is the most difficult part of the genome to assemble. Researchers obtained the gorilla Y chromosome genome by Illumina and PacBio sequencing, with a size of 25.4 Mb with a scaffold N50 of 97.45 Kb and an NG50 of 99.19 kb.
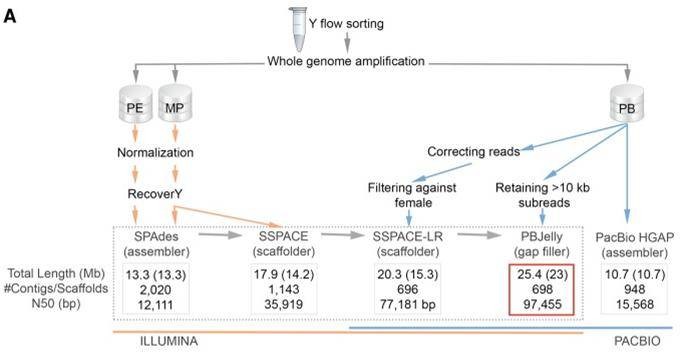 Figure 1. The workflow applied for the Y Chromosome assembly of the gorilla (Tomaszkiewicz M et al., 2016)
Figure 1. The workflow applied for the Y Chromosome assembly of the gorilla (Tomaszkiewicz M et al., 2016)
Similar to the gorilla sex chromosome, the human Y chromosome has a highly readable sequence and is extremely difficult to assemble. The Y chromosome DNA obtained from flow-sorting and enrichment was sequenced by a combination of short-read and long-read sequencing. After assembly, the initial version of the assembly was polished with pilon software using NGS data with higher single-base accuracy, followed by an additional round of Racon error correction, resulting in a Y chromosome genome size of 21.5 Mb. Compared to the gorilla Y chromosome assembly (contig N50=17.95Kb), the continuity was improved by 80-fold (contig N50=1.46Mb).
 Figure 2. Human chromosome-Y assembly (Kuderna L F K et al., 2019)
Figure 2. Human chromosome-Y assembly (Kuderna L F K et al., 2019)
For highly repetitive genomes like mammals, the use of long-read sequencing can greatly simplify sex chromosome assembly and could theoretically be extended to other species.
Sex Chromosome Assembly and Evolution in Birds
In this study, the genomes of five female birds of paradise were sequenced by Illumina and assembled using ALLPATHS-LG. Using the published Z chromosome sequences of the great tit and hooded crow genomes as reference genomes, the sequenced species were compared genome-wide with the reference genomes, and the scaffold on the comparison was classified as a Z chromosome sequence, and the Z chromosomes were assembled. Theoretically, the Z and W chromosomes of female paradise birds should be sequenced at half the depth of autosomes, and the scaffold of the W chromosome should be picked out to complete the assembly of the W chromosome based on the principle.
In the present study, the Z and W sex chromosomes of birds were assembled by genome-wide comparison with species that had completed the sequencing of Z chromosomes, based on the principle of depth halving, which helps to explain the evolution of bird sex chromosomes.
Sex Chromosome Evolution in Asparagus
However, the genes responsible for sex differentiation and their organization patterns are still poorly studied, and the mechanisms of sex determination and the composition of sex chromosomes are still unclear. In this study, authors sequenced a diploid (YY) male garden asparagus individual by Illumina and PacBio, assembled Illumina reads using SOAPdenovo2, Gapcloser gaps, and SSPACE scaffold construction; used PacBio reads by PBjelly2 gaps and scaffold enhancement by PBjelly2; resequencing of 35 XX females and 39 YY supermales, constructing a linkage map, and genome-assisted assembly using genetic and optical mapping, resulting in genome size of 1.3G.
In this study, the diploid (YY) genome of Asparagus was assembled and sex-determining regions were identified. Resequencing analysis showed that dioecious plants have only recently originated in the genus Asparagus and have the same XY morphology; the study of the Y chromosome gene structure and sex-determining mechanism of dioecious plants provides a reference for sex determination in Asparagus.
Summary
Long-read sequencing is expected to achieve complete sequence assembly of sex chromosomes as soon as possible. To date, hundreds of sex chromosomes of plants and animal have been sequenced, assembled and released with varying degrees of continuity and completeness. As genome sequencing technologies continue to advance, high-fidelity long-read sequencing continues to improve, and fractal assembly and scaffolding algorithms improve, it is expected that the assembly of highly contiguous pairs of sex chromosomes will soon become commonplace.
Recommended Services
CD Genomics offers Human Whole Genome PacBio SMRT Sequencing and Whole Genome Sequencing based on Illumina, Nanopore sequencing and PacBio SMRT sequencing platforms, accelerating high-quality sex chromosome genome assembly, evolution and functional research.
You might also be interested in the following articles:
Successful Decoding of the Y Chromosome: A Milestone in Human Genome Unraveling
Chromosome Map by Genome Sequencing Is Allowed to Understand of Chromatin Accessibility at The Genome-wide Level
References:
- Tomaszkiewicz M, Rangavittal S, Cechova M, et al. A time-and cost-effective strategy to sequence mammalian Y Chromosomes: an application to the de novo assembly of gorilla Y. Genome Research, 2016, 26(4): 530-540.
- Kuderna L F K, Lizano E, Julià E, et al. Selective single molecule sequencing and assembly of a human Y chromosome of African origin. Nature communications, 2019, 10(1): 4.
- Xu L, Auer G, Peona V, et al. Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nature ecology & evolution, 2019, 3(5): 834.
- Harkess A, Zhou J, Xu C, et al. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nature communications, 2017, 8(1): 1279.

 Sample Submission Guidelines
Sample Submission Guidelines


