Upon completing the DNA methylation sequencing workflow, the downstream experimental design assumes paramount importance for further validation of specific features of interest within the analysis outcomes. The subsequent validation phase of DNA methylation research encompasses a multitude of experimental approaches. These include foundational and straightforward validation procedures, as well as more intricate whole-genome methylation perturbation experiments (untargeted). Moreover, it extends to targeted methylation/demethylation assays aimed at specific genes to ensure the accuracy and reliability of the study findings.
Validating DNA Methylation Sequencing Results through Simple Methods
Simple validation primarily entails the confirmation of the DNA methylation status and gene expression levels of target genes, involving the following methods:
Verification of Target Gene DNA Methylation: Targeted Bisulfite Sequencing (Target-BS)
Targeted Bisulfite Sequencing (Target-BS) stands as a robust tool for high-precision validation of specific gene regions' DNA methylation status. Herein lie its principal features and applications:
Principle of the Method: Targeted Bisulfite Sequencing leverages bisulfite treatment to convert unmethylated cytosines (C) into uracils (U), while methylated cytosines (5mC) remain unchanged. Through high-throughput sequencing, the distinction between methylated and unmethylated cytosines is achieved.
Ultra-High Depth Sequencing: The sequencing depth of target regions can reach several hundred to thousands of times coverage, ensuring sensitivity and accuracy in detection. For instance, Bibikova et al. (2011) utilized Targeted Bisulfite Sequencing to investigate methylation patterns in breast cancer, demonstrating its efficacy in detecting tumor-associated methylation alterations.
Region Selection and Primer Design: Specific gene regions of less than 300 base pairs need to be chosen, and primers specific for bisulfite treatment are designed for pre-amplification experiments. For example, Gu et al. (2011) successfully designed and validated specific primers targeting multiple critical gene regions while studying mammalian genomic methylation patterns.
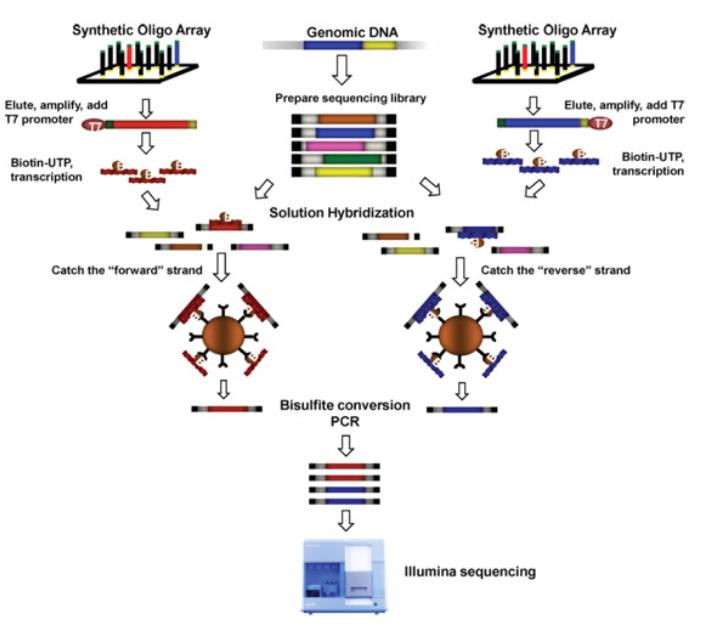 Targeted bisulfite sequencing. Illustrated are the steps involved in the preparation of biotinylated RNA capture probes (top left and right), whole-genome fragment input library (top middle) and hybrid selection-enriched output library (middle left and right). The captured DNA was treated with sodium bisulfite, amplified by PCR and sequenced using an Illumina GAIIx sequencer. (Eun-Joon Lee et al,. 2011)
Targeted bisulfite sequencing. Illustrated are the steps involved in the preparation of biotinylated RNA capture probes (top left and right), whole-genome fragment input library (top middle) and hybrid selection-enriched output library (middle left and right). The captured DNA was treated with sodium bisulfite, amplified by PCR and sequenced using an Illumina GAIIx sequencer. (Eun-Joon Lee et al,. 2011)
Detecting mRNA expression levels of target genes: RT-qPCR
Reverse Transcription Quantitative PCR (RT-qPCR) is a highly sensitive technique utilized for the quantitative measurement of mRNA expression levels, offering precise data support by quantitatively analyzing gene expression changes.
Methodology: mRNA is initially reverse transcribed into cDNA, followed by amplification and detection through quantitative PCR. During the amplification process, fluorescence dyes or probes are employed to monitor the real-time PCR amplification curve. In the realm of DNA methylation research, Yang et al. (2014) utilized RT-qPCR to analyze the transcriptional changes of specific methylated genes, thereby validating the impact of methylation on gene expression.
Data interpretation: Relative expression levels of target genes under different treatment conditions are calculated based on fluorescence signal intensity. Common reference genes (e.g., GAPDH or ACTB) are employed for data standardization.
Protein Immunoblot Analysis (Western Blot)
Western Blotting is a classic technique employed for the detection of target gene protein expression levels, validating the presence and relative abundance of proteins through protein electrophoresis separation and specific antibody recognition.
Methodology: Following SDS-PAGE protein separation, target proteins are transferred onto PVDF or nitrocellulose membranes. Specific antibodies are then utilized for recognition, and signal detection is achieved through chemiluminescence or fluorescence. Towbin et al. (1979) initially proposed the Western Blotting technique, furnishing protein research with a robust tool.
Data interpretation: Relative protein expression levels of target proteins are calculated based on the ratio of target proteins to internal reference proteins (such as β-actin or GAPDH). For instance, Jones et al. (2011) detected the protein expression levels of the methylation-associated gene MGMT implicated in tumorigenesis via Western Blotting, elucidating the regulatory mechanisms of methylation on protein expression. Additionally, Nguyen et al. (2010) investigated the impact of DNA methylation inhibitors on ERα protein expression in breast cancer cells, employing Western Blotting to validate the efficacy of methylation inhibitors.
These straightforward validation methods provide foundational data support for DNA methylation research, aiding in the comprehension of the specific role of gene methylation in gene expression regulation and disease mechanisms.
Genome-wide Untargeted DNA Methylation Interference Experiments
Genome-wide Untargeted DNA methylation interference experiments represent a cornerstone method for studying the impact of DNA methylation on gene expression and cellular function. These experiments involve broad interference with DNA methylation status to explore the role of methylation across the entire genome.
DNA Methylation Interference
Knockdown/Knockout/Overexpression of DNA Methyltransferases
Using genetic engineering techniques such as RNA interference (RNAi), the CRISPR-Cas9 system, or overexpression vectors, interference with the expression or function of DNA methyltransferases (e.g., DNMT1, DNMT3A, DNMT3B) alters the overall methylation status of cells. For instance, Jones et al. (2012) observed a significant reduction in genome-wide methylation levels and subsequent gene expression changes in mouse embryonic stem cells by knocking out the DNMT1 gene. Additionally, Okano et al. (1999) elucidated the crucial roles of these methyltransferases in early mouse development and genomic imprinting through the knockout of DNMT3A and DNMT3B genes.
DNA Methylation Inhibitors
Chemical agents, such as 5-azacytidine (5-Aza), function by forming covalent bonds with DNA methyltransferases, thereby inhibiting their enzymatic activity and reducing cellular DNA methylation levels. Christman et al. (2002) investigated the impact of 5-Aza on DNA methylation and gene expression in cancer cells, revealing its significant ability to decrease genome-wide methylation levels and activate tumor suppressor genes. In a study on breast cancer cells, Glover et al. (1987) treated cells with 5-Aza, resulting in the upregulation of multiple genes, underscoring the pivotal role of DNA methylation in gene silencing.
Detection of Global DNA Methylation Changes
5mC Methylation Immunofluorescence Staining (Qualitative)
Immunofluorescence staining using anti-5mC antibodies enables the visualization of DNA methylation distribution and changes within cells under a fluorescence microscope. Wu et al. (2010) employed 5mC immunofluorescence staining techniques to investigate the methylation status across different cell types, revealing the dynamic alterations of methylation during cellular differentiation.
DNA Spot Hybridization (Qualitative)
DNA samples are spotted onto a membrane, and overall DNA methylation levels are detected through probe hybridization. This method offers a rapid and straightforward means of assessing global methylation levels.
Colorimetric Assay (Quantitative)
The quantification of 5mC content in cell or tissue samples is achieved through enzyme-linked immunosorbent assay (ELISA) or other colorimetric detection methods. Mohn et al. (2008) employed ELISA to quantitatively assess DNA methylation levels in mouse embryonic stem cells, providing precise methylation quantification data.
Mass Spectrometry (Quantitative)
Quantitative analysis of methylation levels in DNA samples is accomplished through liquid chromatography-mass spectrometry (LC-MS) technology. Liu et al. (2007) utilized LC-MS to analyze DNA methylation status in cancer cells, identifying methylation biomarkers associated with tumor progression.
Detection of DNA Methylation Changes in Target Genes: Target-BS
Target-BS is employed to assess changes in the methylation status of specific genes, validating the specific effects of genome-wide interference experiments. This method, through precise region-specific sequencing, provides high-resolution methylation data.
Detection of mRNA Expression Levels of Target Genes: RT-qPCR
RT-qPCR is utilized to analyze the mRNA expression levels of specific genes following methylation interference experiments, elucidating the impact of methylation on gene transcription. This technique, employing reverse transcription and quantitative PCR, offers highly sensitive expression data.
Detection of Protein Expression Levels of Target Genes: Western Blotting
Western Blotting is employed to detect the protein expression levels of specific genes following methylation interference experiments, validating the regulatory role of methylation on protein expression. Through protein separation, transfer, and specific antibody detection, this method provides precise protein expression information.
Through these methods, researchers can comprehensively understand the functional role of DNA methylation in genome regulation, providing essential data support for exploring disease mechanisms and therapeutic targets.
Validation of Targeted Gene Methylation/Demethylation Experiments
Validation of targeted gene methylation/demethylation experiments aims to comprehensively investigate the impact of methylation status in specific gene regions on gene expression and its biological functions. These experiments typically involve precise gene editing and gene expression analysis techniques to ensure the accuracy and reproducibility of results.
Interference of Target Gene DNA Methylation
Luciferase Activity Assay
This experimental approach consists of executing the methylation of plasmids in vitro through the utilization of CpG methyltransferases, thereby assembling target gene-luciferase expression plasmids incorporating both methylated and unmethylated promoters. Upon insertion into the cells, these plasmids, enable the exploration of the effects that methylation of the target gene promoter exerts on gene expression. This is performed by quantifying the luciferase activity. To illustrate, in a pioneering study by Tiwari et al. (2008), the researchers implemented luciferase reporter gene assays and provided compelling evidence demonstrating that methylation of the promoter region of the RUNX3 gene markedly attenuates its expression.
CRISPR-Cas9 Targeted Editing of DNA Methylation Experiment
The CRISPR-Cas9 system stands as a potent genetic editing tool, capable of introducing or removing methylation modifications at specific DNA sequences by combining dCas9 (deactivated Cas9) with methyltransferases (such as DNMT3A) or demethylases (such as TET1). This technology finds extensive application in targeted research on gene-specific methylation/demethylation. For instance, Liu et al. (2016) successfully targeted methylation to the promoter region of the p16INK4a gene in human cells using the CRISPR-dCas9-DNMT3A system, resulting in a significant reduction in its expression.
Detection of DNA Methylation Changes in Target Genes
Target-BS
This method not only identifies methylation sites but also quantitatively analyzes the degree of methylation. For instance, Kulis et al. (2012), utilizing Target-BS technology, conducted a detailed analysis of methylation patterns in critical gene regions among patients with chronic lymphocytic leukemia.
Contrasting Whole-Genome Bisulfite Sequencing (WGBS) with TBS: An Epigenetic Analysis Perspective
In the realm of epigenetic analysis, the comparison between Whole-Genome Bisulfite Sequencing (WGBS) and TBS finds resonance with the distinction between RNA-seq and RT-qPCR in gene expression studies. Here's a detailed exploration of this analogy:
Comprehensive Genome-wide Coverage vs. Targeted Precision
Much like RNA-seq offers a comprehensive view of gene expression across the entire transcriptome, WGBS provides an expansive assessment of DNA methylation patterns throughout the entire genome. In contrast, TBS, analogous to RT-qPCR, offers targeted precision by focusing on specific genomic regions of interest, enabling researchers to delve deep into the methylation dynamics of select loci.
Depth of Coverage: High-Resolution vs. Ultra-High Precision
WGBS, akin to RNA-seq's high-resolution profiling, generates extensive data by sequencing DNA fragments from bisulfite-treated genomic DNA across the entire genome. This approach yields valuable insights into methylation patterns at various genomic scales. Conversely, TBS, reminiscent of the ultra-high precision of RT-qPCR, achieves remarkable sequencing depth, often reaching several hundred to thousands of times coverage. This depth ensures robust and reliable methylation profiling at single-base resolution within the targeted regions.
Scalability and Flexibility
While WGBS offers scalability and flexibility by surveying the entire genome, TBS provides flexibility in experimental design, allowing researchers to tailor their analyses to specific genomic regions of interest. This targeted approach reduces sequencing costs and computational resources, making it an efficient strategy for studying methylation dynamics in defined genomic loci.
Customization and Experimental Design
In both RNA-seq and RT-qPCR, experimental design and primer selection are critical for obtaining accurate and meaningful results. Similarly, TBS requires careful primer design for bisulfite conversion and subsequent amplification of target regions. This customization ensures the specificity and sensitivity necessary for accurately assessing DNA methylation status in the desired genomic loci.
By drawing parallels between WGBS and RNA-seq, as well as TBS and RT-qPCR, researchers can gain a nuanced understanding of the strengths and applications of these epigenetic analysis techniques. Whether exploring genome-wide methylation patterns or investigating specific regulatory regions, the choice between WGBS and TBS depends on the research objectives, resources, and desired level of precision.
You may interested in
Learn More
Detection of mRNA Expression Levels of Target Genes: Real-Time Quantitative PCR (RT-qPCR)
Huang et al. (2011) utilized RT-qPCR to evaluate the changes in mRNA expression levels of the BRCA1 gene in breast cancer cells following treatment with demethylating agents. Their study demonstrated the restoration of gene expression upon demethylation treatment.
Detection of Protein Expression Levels of Target Genes: Western Blotting
Esteller et al. (2000) employed Western Blot analysis to measure the protein expression levels of the hMLH1 gene in colorectal cancer cells after treatment with demethylating agents. The study confirmed the restoration of protein expression following demethylation treatment.
Assessment of Cellular Functional Impacts
Immunofluorescence Microscopy Observation
Using immunofluorescence staining and microscopic observation, researchers analyzed the impact of changes in target gene expression on cell morphology and function.
Functional Biomarker Assays
By detecting biomarkers related to cell cycle, proliferation, apoptosis, and other functions, the influence of target gene methylation status changes on cellular function is assessed. For instance, Good et al. (2017) investigated the role of TET2 in acute myeloid leukemia (AML) and demonstrated its crucial involvement in cell proliferation and apoptosis through functional biomarker analysis. Their research revealed that TET2 mutations, by affecting DNA demethylation levels, lead to aberrant proliferation and anti-apoptotic properties in AML cells. These findings underscore the significance of TET2 in regulating cellular function and emphasize DNA methylation as a potential therapeutic target in cancer.
References:
- Jones, P. A., et al. (2011). "Targeting the cancer epigenome for therapy." Nature Reviews Genetics, 12(9), 631-643.
- Nguyen, T. T., et al. (2010). "Role of estrogen receptor alpha in breast cancer development and progression: from clinical evidence to molecular insights." Oncology Reports, 24(4), 1193-1201.
- Bibikova, M., et al. (2011). "High density DNA methylation array with single CpG site resolution." Genomics, 98(4), 288-295. Gu, H., et al. (2011). "Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution." Nature Methods, 7, 133-136.
- Schmittgen, T. D., & Livak, K. J. (2008). "Analyzing real-time PCR data by the comparative C(T) method." Nature Protocols, 3(6), 1101-1108.
- Livak, K. J., & Schmittgen, T. D. (2001). "Analysis of relative gene expression data using real-time quantitative PCR and the 2^(-Delta Delta C(T)) Method." Methods, 25(4), 402-408.
- Towbin, H., et al. (1979). "Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications." Proceedings of the National Academy of Sciences, 76(9), 4350-4354.
- Burnette, W. N. (1981). "'Western blotting': electrophoretic transfer of proteins from sodium dodecyl sulfate–polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A." Analytical Biochemistry, 112(2), 195-203.
- Jones, P. A., & Liang, G. (2012). "A decade of exploring the cancer epigenome - biological and translational implications." Nature Reviews Cancer, 12(3), 147-160.
- Okano, M., Bell, D. W., Haber, D. A., & Li, E. (1999). "DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development." Cell, 99(3), 247-257.
- Christman, J. K. (2002). "5-Azacytidine and 5-aza-2'-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy." Oncogene, 21(35), 5483-5495.
- Glover, T. W., et al. (1987). "DNA methylation and cancer." In: Feinberg AP, et al. (eds) DNA Methylation. Basic Mechanisms and Applications. CRC Press, Boca Raton.
- Mohn, F., et al. (2008). "Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors." Molecular Cell, 30(6), 755-766.
- Liu, S., et al. (2007). "High-throughput DNA methylation analysis by liquid chromatography/tandem mass spectrometry." Clinical Chemistry, 53(3), 658-664.
- Tiwari, V. K., et al. (2008). "PcG proteins, DNA methylation, and gene repression by chromatin looping." PLoS Biology, 6(12), e306. Liu, X. S., et al. (2016). "Editing DNA methylation in the mammalian genome." Cell, 167(1), 233-247.
- Kulis, M., et al. (2012). "Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia." Nature Genetics, 44(11), 1236-1242.
- Huang, T. H. M., et al. (2011). "Epigenetic gene silencing in cancer: mechanisms and clinical implications." Epigenomics, 3(1), 73-90. DOI: 10.2217/epi.10.69
- Esteller, M., et al. (2000). "MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas." Oncogene, 19(10), 1376-1383.
- Choi, S. H., et al. (2017). "DNMT3B is required for the growth and survival of non-small cell lung cancer cells independently of its DNA methyltransferase activity." Oncotarget, 8(9), 15336-15348.
- Good, C., et al. (2017). "TET2 mutations affect non-CpG island DNA methylation at enhancers and transcription factor-binding sites in chronic myelomonocytic leukemia." Cancer Cell, 31(5), 660-675.

 Sample Submission Guidelines
Sample Submission Guidelines


