DNA Methylation
DNA Methylation, a fundamental biological process, involves the addition of a methyl group to specific bases within the DNA sequence through covalent bonding facilitated by DNA methylation transferase (DNMT).
This chemical modification is a pivotal form of epigenetic regulation, preserving DNA sequence while influencing gene activity. It exerts profound effects on various biological phenomena including gene expression, embryonic development, cell proliferation, differentiation, genome stability maintenance, and defense against exogenous DNA invasions.
DNA methylation occurs at different positions on DNA bases such as the C-5 position of cytosine, N-6 position of adenine, and N-7 position of guanine, catalyzed by various DNA methylation enzymes. Notably, the methylation of the 5th carbon atom of cytosine at CpG sites (cytosine-phosphate-guanine sites) is extensively researched, yielding 5-methylcytosine (5-mC).
5-mC, the resultant product, is ubiquitous across the genomes of plants, animals, and other eukaryotic organisms, representing one of the most extensively studied forms of DNA methylation modification.
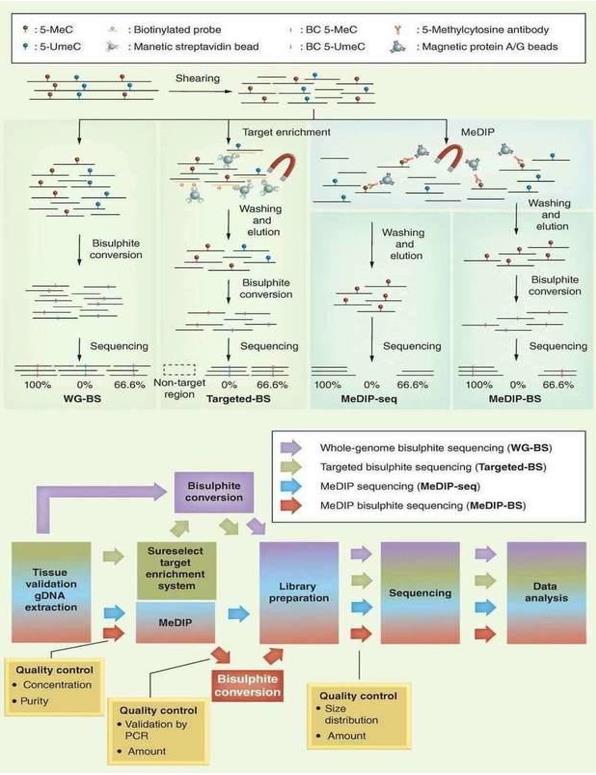 Different NGS-based DNA methylation analysis methods. (Jeong et al., 2016)
Different NGS-based DNA methylation analysis methods. (Jeong et al., 2016)
What is Bisulfite Sequencing (BS-seq)?
Bisulfite sequencing, often abbreviated as BS-seq, is a powerful method used to detect DNA methylation patterns at single-base resolution. By treating DNA with bisulfite, unmethylated cytosines are converted to uracil, while methylated cytosines remain unchanged. This differential conversion allows researchers to distinguish between methylated and unmethylated cytosines when analyzing the DNA sequence. This technique is crucial for understanding epigenetic regulation and its role in various biological processes, including development, disease, and gene expression.
DNA methylation information may be lost during standard molecular biology manipulations. CD Genomics offers different sequencing platforms that facilitate the robust analysis of genome-wide epigenomics. This advanced sequencing approach allows for comprehensive and efficient examination of DNA methylation, providing valuable insights into the molecular landscape and potential biomarkers associated with various conditions.
The principle of whole genome BS-seq involves treating genomic DNA with sodium bisulfite, which converts all unmethylated cytosines to uracil while leaving methylated cytosines unchanged. After bisulfite treatment, primers are designed to amplify regions of interest, typically CpG islands, via PCR. The resulting PCR products are then purified and cloned using TA cloning. Positive clones are selected and subjected to sequencing, allowing for the determination of the methylation status at each CpG site. Finally, the sequenced data is aligned with the original genomic sequence to determine the number and location of methylated sites, as well as to analyze the overall methylation level.
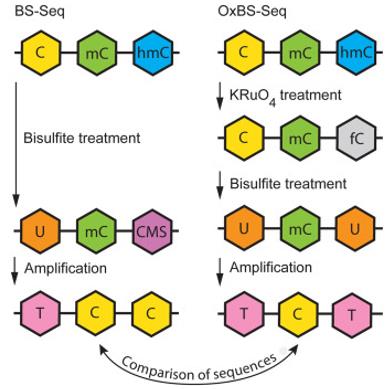 Oxidative bisulfite sequencing (OxBS-seq) and standard bisulfite sequencing (BS-seq). (Rauch et al., 2023)
Oxidative bisulfite sequencing (OxBS-seq) and standard bisulfite sequencing (BS-seq). (Rauch et al., 2023)
The Workflow of BS-seq
- Quality Testing of DNA Samples
DNA samples undergo initial quality assessment to ensure suitability for sequencing.
Genomic DNA is fragmented into 100-300bp fragments via sonication.
DNA ends are repaired, an A base is added at the 3' end, and sequencing adapters are ligated.
Bisulfite treatment is applied to convert unmethylated cytosines to uracil.
Desalting and gel purification steps are performed to select appropriate library fragment sizes.
PCR amplification is conducted to enrich library fragments, followed by another round of size selection.
Quality control checks are conducted on the constructed libraries.
Libraries passing quality control are subjected to high-throughput sequencing.
Sequencing results are aligned to the reference genome.
Unique sequences are extracted for subsequent analysis.
Initial data filtering is performed to remove low-quality reads.
Usable data quantity is evaluated to ensure compliance with project requirements.
Comparison of available data with the reference genome is conducted, yielding comparison results.
Quality-checked comparison data is utilized to derive genome-wide methylation information.
Information analysis and processing are carried out to generate standard and personalized analysis results.
Result Interpretation: Methylation patterns and variations are interpreted in the context of biological significance and potential implications for the studied samples.
Advantages of BS-seq
- Provides comprehensive coverage of CpG and non-CpG methylation throughout the genome at single-base resolution.
- Enables analysis of methylation patterns in diverse genomic regions, including dense, less dense, and repetitive sequences.
Challenges of BS-seq
- Bisulfite treatment converts unmethylated cytosines to thymidine, reducing sequence complexity and making it challenging to generate accurate comparisons.
- Potential occurrence of nucleotide positions (NPs) where cytosine conversion to thymidine is incomplete during bisulfite treatment.
- Unable to distinguish between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), leading to ambiguity in methylation status determination.
As technology has advanced, numerous variations of bisulfite sequencing (BS-seq) have emerged, each with its own advantages and applications.
- oxBS-seq (Oxidized Bisulfite Sequencing): As mentioned earlier, oxBS-seq involves oxidizing 5-hydroxymethylcytosine (5hmC) to 5-formylcytosine (5fC) prior to bisulfite treatment, allowing for the precise detection of 5-methylcytosine (5mC).
- TAB-seq (Tet-Assisted Bisulfite Sequencing): This method involves the use of Tet enzymes to oxidize 5mC to 5hmC or further oxidized forms before bisulfite treatment, enabling the discrimination between different cytosine modifications.
- CMS-seq (Chromatin Methylation Sequencing): CMS-seq combines bisulfite sequencing with chromatin immunoprecipitation (ChIP), allowing for the simultaneous analysis of DNA methylation and histone modifications.
- BSPP-seq (Bisulfite Padlock Probes Sequencing): This method utilizes padlock probes to capture target regions of interest before bisulfite treatment, enabling targeted bisulfite sequencing with enhanced coverage and sensitivity.
- BS-PCR (Bisulfite Polymerase Chain Reaction): This technique involves bisulfite treatment followed by PCR amplification of specific target regions, allowing for targeted analysis of DNA methylation.
- BSAS (Bisulfite Amplicon Sequencing): Similar to BS-PCR, BSAS involves bisulfite treatment and PCR amplification of specific target regions, but typically utilizes next-generation sequencing for high-throughput analysis.
Whole Genome Bisulfite Sequencing (WGBS)
Whole Genome Bisulfite Sequencing (WGBS) is a powerful technique used for genome-wide DNA methylation analysis. It provides single-base resolution of DNA methylation across the entire genome.
In WGBS, genomic DNA is treated with sodium bisulfite, which converts unmethylated cytosines to uracil, while methylated cytosines remain unchanged. After bisulfite treatment, high-throughput Next-Generation Sequencing (NGS) is used to sequence the treated DNA fragments. By comparing the sequencing reads to a reference genome, researchers can determine the methylation status of individual cytosines throughout the entire genome.
WGBS has been widely used in various species including humans, plants, animals, and lower organisms to study genome-wide DNA methylation patterns. It provides valuable insights into the role of DNA methylation in gene regulation, development, disease, and evolution.
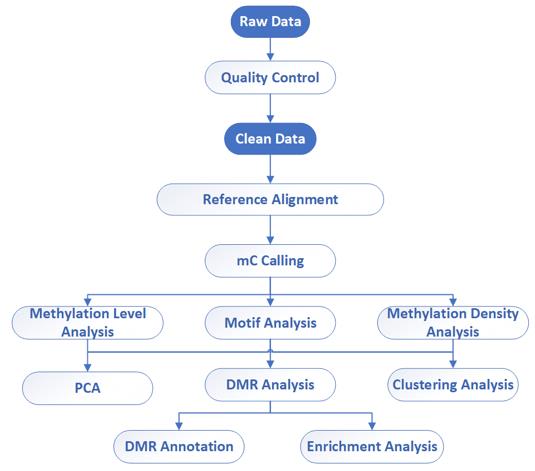 Whole Genome Bisulfite Sequencing (WGBS) – CD Genomics
Whole Genome Bisulfite Sequencing (WGBS) – CD Genomics
Reduced Representation Bisulfite Sequencing (RRBS)
Reduced representation bisulfite sequencing (RRBS) is a method that involves enriching for CCGG-rich fragments in genomic DNA through restriction enzyme digestion. Subsequently, these enriched fragments, which typically contain CpG-rich regions of the genome, undergo single-base resolution methylation sequencing via bisulfite treatment and high-throughput sequencing technology. Compared to Whole Genome Bisulfite Sequencing (WGBS), RRBS is a more cost-effective approach as it requires significantly less sequencing volume while still providing valuable methylation sequencing data. This makes RRBS particularly suitable for large-scale clinical studies involving genome-wide methylation analysis.
In simplified terms, bisulfite treatment converts unmethylated cytosines (C) to uracil (U), which are then read as thymine (T) during sequencing. By comparing the number of reads that are converted to thymine with the total number of reads covering a specific cytosine site, researchers can calculate the methylation rate at that site. This technique is invaluable for studying various biological processes such as embryonic development, aging mechanisms, disease development, and the identification of disease-related epigenetic marker loci.
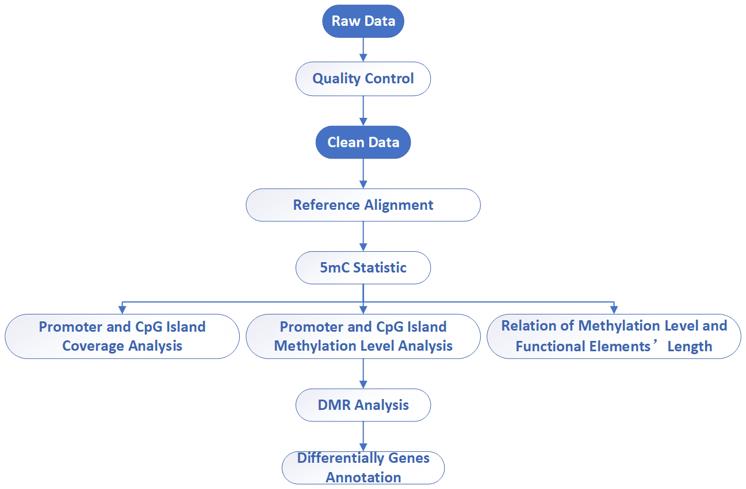 Reduced Representation Bisulfite Sequencing – CD Genomics
Reduced Representation Bisulfite Sequencing – CD Genomics
Oxidative Bisulfite Sequencing (oxBS-seq)
The significance of hydroxymethylation (5hmC) in the mammalian genome and its implications in various biological processes such as development, aging, neurodegenerative diseases, and tumorigenesis. Hydroxymethylation is indeed a relatively recent discovery in the field of epigenetics, and it has garnered significant attention due to its emerging roles and potential implications.
One of the key challenges in studying DNA hydroxymethylation is distinguishing it from DNA methylation (5mC), particularly using conventional bisulfite sequencing methods. As you mentioned, bisulfite treatment converts both 5mC and 5hmC to similar products, making it difficult to differentiate between the two modifications.
Oxidative bisulfite sequencing (oxBS-Seq) is a sophisticated technique that addresses this challenge. By chemically oxidizing 5hmC to a different intermediate product before bisulfite treatment, oxBS-Seq allows for the specific detection of 5mC while excluding the effects of 5hmC. Additionally, oxBS-Seq can be combined with other sequencing approaches to simultaneously detect both DNA methylation and hydroxymethylation at single-base resolution.
This advancement in technology has greatly enhanced our ability to dissect the complex interplay between DNA methylation and hydroxymethylation and their roles in various biological processes and diseases. oxBS-Seq holds immense promise for furthering our understanding of epigenetic regulation and its implications for human health and disease.
BSAS (Bisulfite Amplicon Sequencing)
Bisulfite amplicon sequencing is a targeted approach for analyzing DNA methylation or hydroxymethylation patterns in specific genomic regions of interest. Here's how the technique typically works:
- Primer Design: Methylation-specific amplification primers are designed to target specific DNA sequences within the genome. These sequences often correspond to regions of interest, such as genes or regulatory elements.
- Bisulfite Treatment: Genomic DNA is treated with bisulfite, which converts unmethylated cytosines to uracil (and subsequently thymine during PCR), while methylated cytosines remain unchanged. In the case of hydroxymethylation analysis, an additional oxidation step may be included to convert hydroxymethylcytosine (5hmC) to a different intermediate before bisulfite treatment.
- PCR Amplification: The bisulfite-treated DNA is then subjected to PCR amplification using the methylation-specific primers. This amplification step selectively targets the regions of interest, resulting in the generation of DNA fragments corresponding to those regions.
- Sequencing: The amplified DNA fragments are then subjected to high-throughput sequencing using next-generation sequencing technologies, such as Illumina sequencing. This allows for the accurate detection of the methylation or hydroxymethylation status of the targeted gene regions at single-base resolution.
By focusing on specific genomic regions of interest, bisulfite amplicon sequencing provides a cost-effective and efficient method for targeted DNA methylation or hydroxymethylation analysis. This approach is particularly useful when studying the methylation status of specific genes or regulatory elements associated with particular biological processes or diseases.
Tet-Assisted Bisulfite Sequencing
TAPS sequencing, or Tet-Assisted Bisulfite Sequencing, is an innovative method for analyzing DNA methylation patterns that offers several advantages over traditional bisulfite sequencing techniques.
In Tet-assisted bisulfite sequencing, bisulfite conversion is replaced with a different chemical approach that directly converts methylated cytosines (5mC) to thymine (T) for sequencing. This technique utilizes a combination of enzymatic and chemical reactions to achieve the conversion of cytosine to thymine.
Here's how Tet-assisted bisulfite sequencing works:
- Oxidation of 5mC and 5hmC: Initially, 5mC and 5hmC are oxidized to 5caC (5-carboxycytosine) using TET1 oxidase.
- Conversion to DHU: The oxidized 5caC is then converted to dihydrouracil (DHU) in the presence of the reducing agent pyridine borane (pyridine). DHU can serve as a template during PCR amplification.
- PCR Amplification: During PCR amplification, the DNA polymerase recognizes DHU as uracil (U), leading to the incorporation of adenine (A) opposite the DHU site. As a result, cytosines in the original DNA sequence are effectively converted to thymines in the final PCR product.
The advantages of TAPS technology include:
- Non-destructive to DNA: Unlike bisulfite conversion, Tet-assisted bisulfite sequencing does not degrade DNA, leading to less DNA loss during the process.
- Improved sequencing data quality: Tet-assisted bisulfite sequencing preserves the base balance of the DNA sequence, resulting in higher-quality sequencing data with increased coverage and complexity.
- Cost-effectiveness: Tet-assisted bisulfite sequencing technology is generally more cost-effective than traditional bisulfite conversion methods.
- Additionally, Tet-assisted bisulfite sequencing allows for the retention of longer DNA fragments (up to 10kb), which is beneficial for downstream applications such as triple sequencing.
Overall, Tet-assisted bisulfite sequencing offers a promising alternative to bisulfite sequencing for DNA methylation analysis, providing improved data quality, reduced DNA loss, and cost-effectiveness.
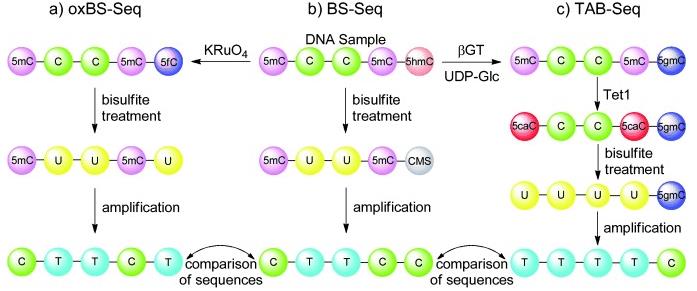 OxBS-Seq, BS-Seq and TAB-Seq. (Schüler et al., 2012)
OxBS-Seq, BS-Seq and TAB-Seq. (Schüler et al., 2012)
Nanopore Sequencing
Nanopore sequencing is a revolutionary technology that enables the direct, real-time sequencing of DNA and RNA molecules.
- Nanopore Detection: Nanopore sequencing involves passing a DNA or RNA molecule through a tiny biological or solid-state nanopore. As the molecule passes through the nanopore, it causes disruptions in the electric current flowing through the pore. These disruptions are characteristic of the specific nucleotide sequence of the molecule.
- Signal Detection: The disruptions in the electric current are detected and recorded as "squiggles" by the nanopore sequencing device. Each nucleotide passing through the pore produces a distinct squiggle pattern, allowing for the determination of the sequence.
- Methylation Detection: In addition to the nucleotide sequence, nanopore sequencing can also detect epigenetic modifications such as DNA methylation. The presence of methylated bases alters the electrical properties of the nucleotides, resulting in unique signal patterns when they pass through the nanopore.
- Deep Learning Models: Deep learning algorithms are employed to analyze the complex signal patterns generated by nanopore sequencing. By training these models on known methylation patterns, researchers can develop tools that accurately identify and characterize methylation modifications in DNA and RNA molecules based on nanopore sequencing signals.
Overall, nanopore sequencing coupled with deep learning models offers a powerful and versatile approach for the direct detection of DNA and RNA sequences, as well as epigenetic modifications such as DNA methylation. This technology has wide-ranging applications in genomics, epigenetics, and biomedical research.
References:
- Rauch, Tibor A., and Gerd P. Pfeifer. "Methods for analyzing DNA cytosine modifications genome-wide." Handbook of Epigenetics. Academic Press, 2023. 123-135.
- Schüler, Peter, and Aubry K. Miller. "Sequencing the Sixth Base (5‐Hydroxymethylcytosine): Selective DNA Oxidation Enables Base‐Pair Resolution." Angewandte Chemie International Edition 51.43 (2012): 10704-10707.
- Jeong H.M., et al. Efficiency of methylated DNA immunoprecipitation bisulphite sequencing for whole-genome DNA methylation analysis. Epigenomics. 2016, 8(8):1061-77.

 Sample Submission Guidelines
Sample Submission Guidelines


