
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

DNA methylation refers to the addition of a methyl group (CH3) to the DNA strand which is catalyzed by DNA methyltransferases (DNMTs). DNA contains four kinds of nitrogenous bases, namely cytosine, guanine, thymine and adenine (Figure 1A). Researchers have found that cytosine and adenine can be methylated. Cytosine methylation often happens at the 5-carbon position of cytosine (5-methylcytosine or 5mC), which is found exclusively at symmetric CG sites on the DNA double helix across the entire genome, namely the CpG island (Figure 2B). Cytosine methylation is widespread in both eukaryotes and prokaryotes (Cloney, 2016; Cooper, 1983). Adenine methylation happens at the 6-nitrogen position of adenine (N6-methyladenine or N6mA). Adenine methylation has been observed in bacterial, plant and mammalian DNA (Ratel et al., 2006; Wu et al., 2016), but has received considerably less attention (Figure 2B).
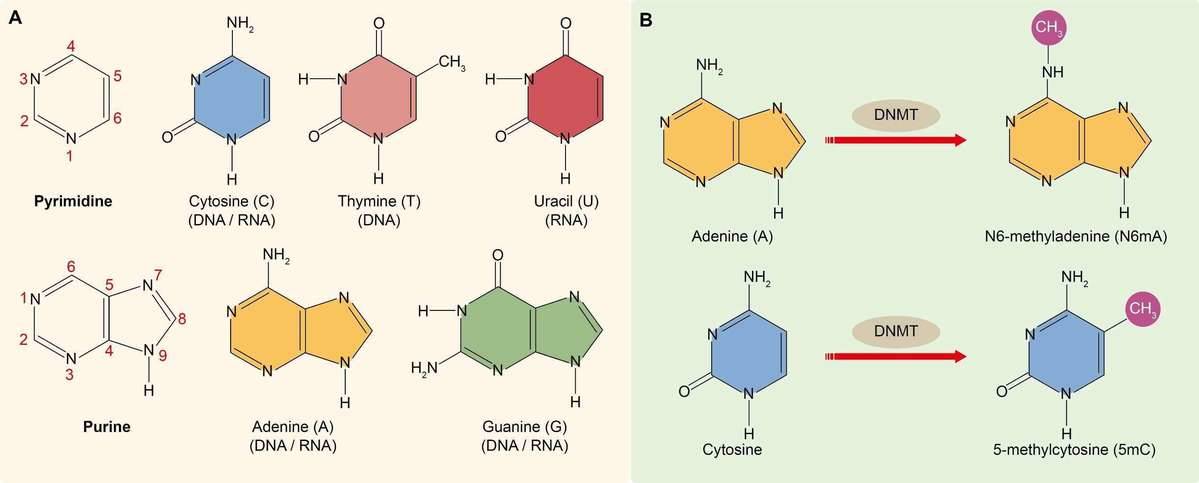 Figure 1. (A) Nitrogenous bases found in DNA and RNA. (B) The methylation of cytosine and adenine.
Figure 1. (A) Nitrogenous bases found in DNA and RNA. (B) The methylation of cytosine and adenine.
DNA methylation is one of the most important epigenetic modifications that is correlated with gene repression (Figure 2) and is known to play an essential role in embryonic development, maintenance of pluripotency, genomic imprinting and X-chromosome inactivation through regulation of transcription, chromatin structure and chromosome stability (Robertson, 2005). DNA methylation can also affect health resulting in cancer, autoimmune disease, neurological disorders or other diseases. In many disease processes, gene promoter CpG islands acquire abnormal hypermethylation, which results in transcriptional silencing that can be inherited by daughter cells following cell division (Wang and Lei, 2018). Alterations of DNA methylation patterns have been recognized as an important component of cancer development (Figure 3).
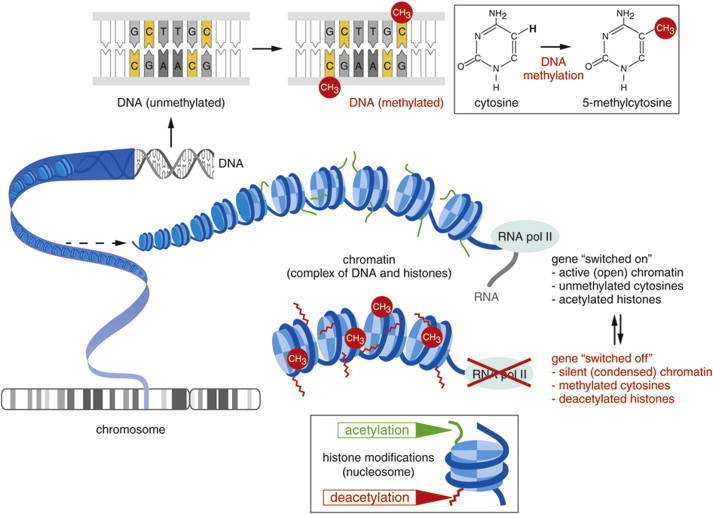 Figure 2. Gene expression can be regulated before the initiation of transcription by the chemical modification of DNA or associated histone proteins (DNA methylation and histone acetylation) (Mukherjee et al., 2015).
Figure 2. Gene expression can be regulated before the initiation of transcription by the chemical modification of DNA or associated histone proteins (DNA methylation and histone acetylation) (Mukherjee et al., 2015).
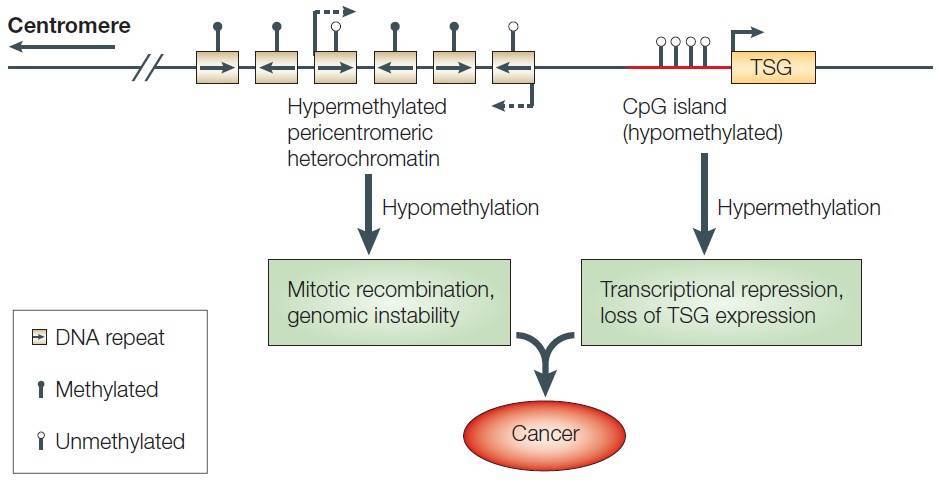 Figure 3. DNA methylation and cancer. TSG: tumor suppressor gene. (Robertson, 2005)
Figure 3. DNA methylation and cancer. TSG: tumor suppressor gene. (Robertson, 2005)
DNA Methylation Detection and RRBS
Understanding the role of DNA methylation in development and disease requires knowledge of the distribution of these modifications in the genome (Harris et al., 2010). Measuring the total amount of 5mC or 5hmC (5-hydroxymethylcytosine) allows researchers to gain insight into profound biological processes and identify biomarkers for disease. The detection of DNA methylation patterns is a rapidly advancing area of research and several methods have been available for the assessment of DNA methylation, with bisulfite treatment being a central procedure to a majority. A simple algorithm for choosing a suitable method for DNA methylation analysis is illustrated in Figure 4 (Kurdyukov and Bullock, 2016).
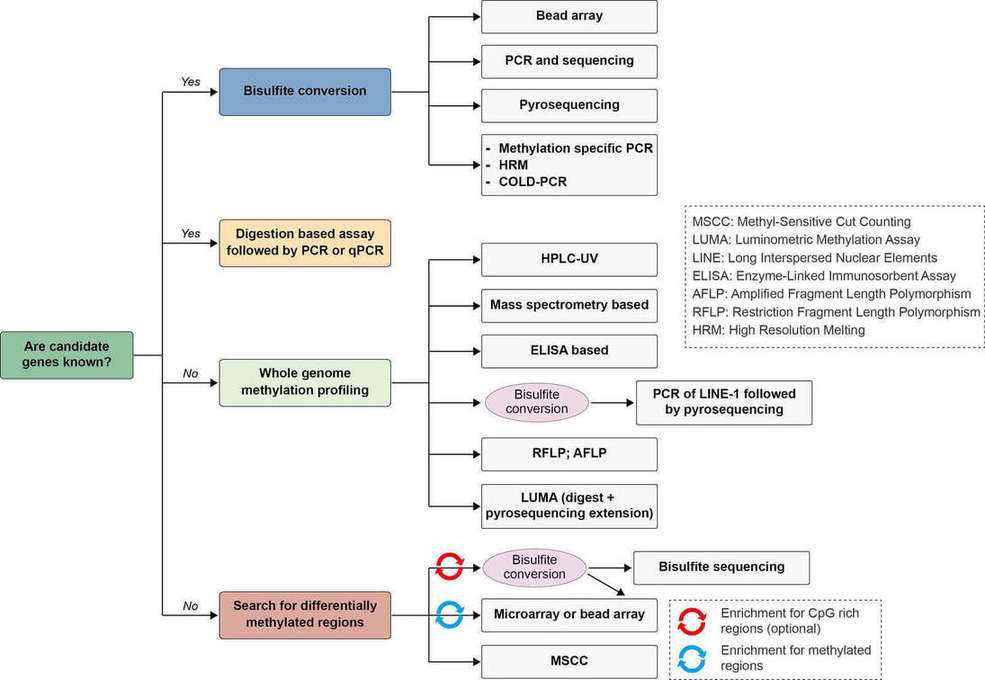 Figure 4. Algorithm for choosing a suitable method for DNA methylation analysis. Adapted from (Kurdyukov and Bullock, 2016)
Figure 4. Algorithm for choosing a suitable method for DNA methylation analysis. Adapted from (Kurdyukov and Bullock, 2016)
Bisulfite sequencing is the use of bisulfite treatment of DNA to determine its pattern of methylation (Frommer et al., 1992). The bisulfite treatment of DNA mediates the deamination of cytosine into uracil, and these converted residues will be read as thymine, as determined by PCR-amplification and subsequent sequencing analysis (Figure 5).
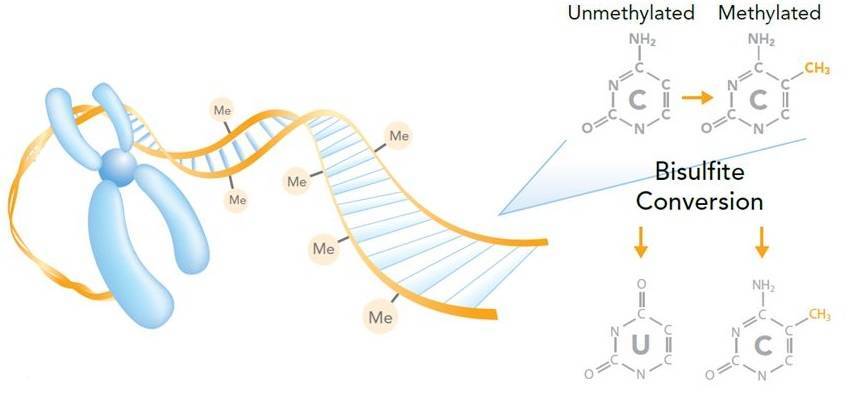 Figure 5. Measuring DNA methylation by bisulfite sequencing (Imaged by Illumina).
Figure 5. Measuring DNA methylation by bisulfite sequencing (Imaged by Illumina).
Whole genome bisulfite sequencing (WGBS) is the most comprehensive method of DNA methylation analysis, and now has been considered as the “gold standard” method in DNA methylation studies. The only limitations are the cost and difficulties in the analysis of NGS data. Reduced representation bisulfite sequencing (RRBS), which combines restriction enzymes and bisulfite sequencing to enrich for areas of the genome with a high CpG content, is an efficient and high-throughput technique for analyzing the genome-wide methylation profiles on a single nucleotide level. The method can reduce the number of nucleotides required to sequence to 1% of the genome (Meissner et al., 2005). Due to its economical and productive manner, RRBS has been used in quickly profiling of aberrant methylation in cancer (Smith et al., 2009) and methylation states in development.
Workflow of RRBS
The protocol of RRBS is illustrated in Figure 6. The process consists of several steps:
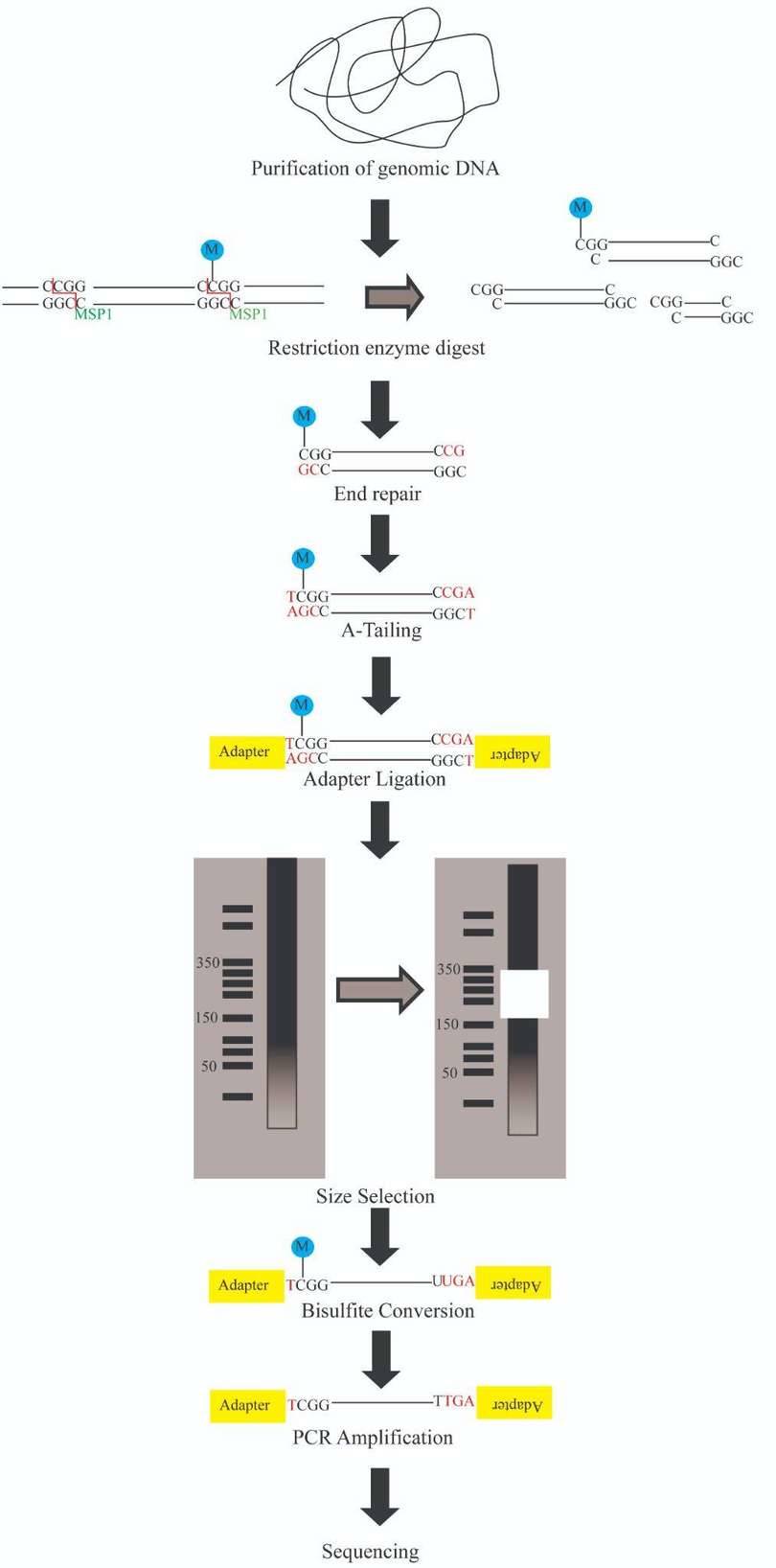 Figure 6. Reduced Representation Bisulfite Sequencing Protocol. (from google)
Figure 6. Reduced Representation Bisulfite Sequencing Protocol. (from google)
Advantages and Limitations of RRBS
RRBS has some technical advantages over WGBS and other DNA methylation detection methods. But there are also some limitations in the specific protocol steps.
At CD Genomics, we are dedicated to providing reliable epigenomics sequencing services, including EpiTYPER DNA methylation analysis, targeted bisulfite sequencing, reduced representation bisulfite sequencing (RRBS), whole genome bisulfite sequencing, MeDIP sequencing, ChIP-seq, and MethylRAD-seq.
References:


