Genome-wide analysis, based on whole genome sequencing, refers to the comprehensive and systematic study of an organism's entire genetic information. With the rapid development of high-throughput sequencing technologies, genome-wide analysis has become a core tool in biological research. In the field of plant science, this technology enables us to gain in-depth insights into the composition, function, and variations of plant genomes under different environmental conditions. Plant genome-wide analysis not only reveals the structure and function of plant genomes but also provides crucial information for crop improvement, disease resistance studies, environmental adaptation, and more.
What is Plant Genomes
A plant genome encompasses all the genetic material within a plant, including its genome, transcriptome, epigenome, and other omics data. Compared to animals and microorganisms, plant genomes vary widely in size. The genome size of plants is closely related to factors such as species complexity, reproductive strategies, and environmental conditions. For example, crop genomes like those of wheat and rice are relatively large, while the genomes of model plants like Arabidopsis thaliana are much smaller.
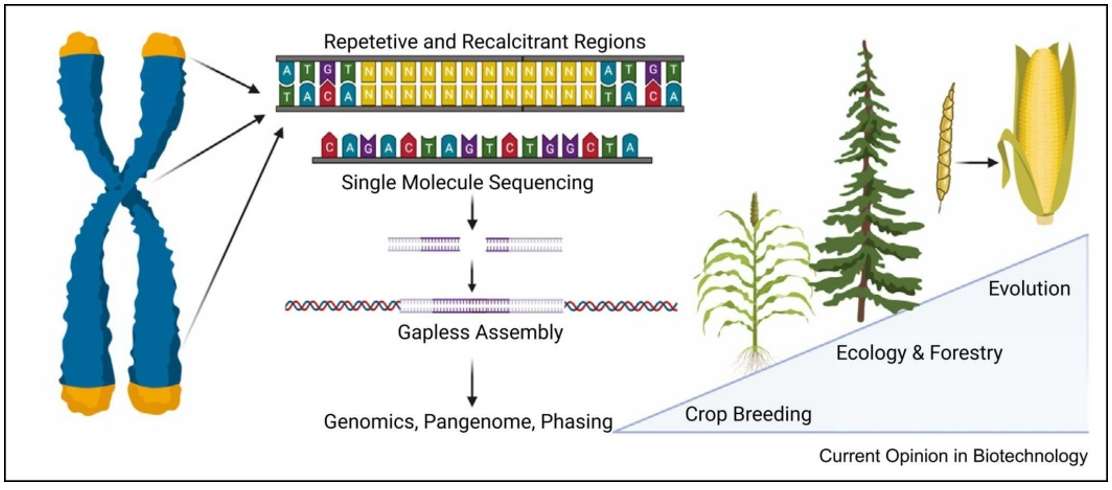 Fig. 1. Era of gapless plant genomes.(Gladman., et.al, 2021).
Fig. 1. Era of gapless plant genomes.(Gladman., et.al, 2021).
The plant genome typically consists of exons, introns, transcribed regions, and non-coding regions. It also contains a significant proportion of repetitive sequences and transposable elements, which play a key role in genome stability, evolution, and species adaptation. Moreover, plant genomes exhibit considerable diversity across species, reflected in variations in gene family sizes, functional diversity, and epigenetic differences.
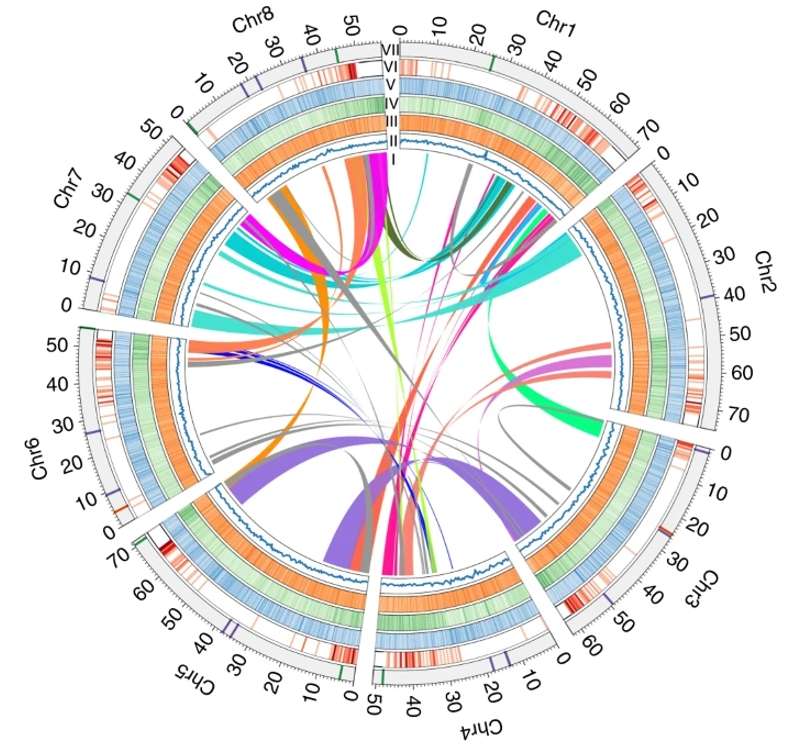 Fig. 2. A classic genome structure in plant.(Li, M., et.al, 2019).
Fig. 2. A classic genome structure in plant.(Li, M., et.al, 2019).
Technological Foundations of Genome-Wide Analysis
In recent years, the advancement of high-throughput sequencing technologies has greatly enhanced the efficiency and precision of genome-wide analysis. The Illumina platform provides cost-effective, high-accuracy short-read sequencing, while PacBio and Oxford Nanopore offer long-read sequencing capabilities that help overcome challenges related to repetitive regions and structural variations. These technologies enable researchers to assemble and annotate plant genomes at a high quality.
Genome assembly is the first step in genome analysis, where short reads are stitched together into longer continuous fragments (contigs), followed by chromosome-level assembly. Genome annotation then helps to determine the location, function, and relationships of genes. In addition to the genome itself, other omics techniques like transcriptomics, epigenomics, and metabolomics play an essential role in providing a more comprehensive genome-wide view. By integrating data from these different omics layers, a fuller understanding of plant genomics can be achieved.
Analysis and Processing of Plant Genome Data
The analysis of plant genome data is a complex multi-step process. Initially, raw sequencing data undergo quality control and preprocessing to remove low-quality sequences, ensuring data accuracy. Next, genome assembly stitches short-read sequences together, building a draft of the genome, which is followed by annotation to identify genes and their functions.
Genome Feature Assessment and Assembly of Simple Genomes
Genome size, heterozygosity, and repeat content are critical factors influencing sequencing and assembly. K-mer distribution analysis can evaluate basic genome characteristics, such as sequencing errors, heterozygosity, and repeat proportions. Simple genomes (homozygous or low heterozygosity) are typically assembled by combining second- and third-generation sequencing, leveraging the long-read advantage of third-generation data to enhance assembly quality. High-quality assembly relies on constructing chromosome-level genomes using genetic maps or Hi-C maps, with sequence error correction being essential for improving accuracy.
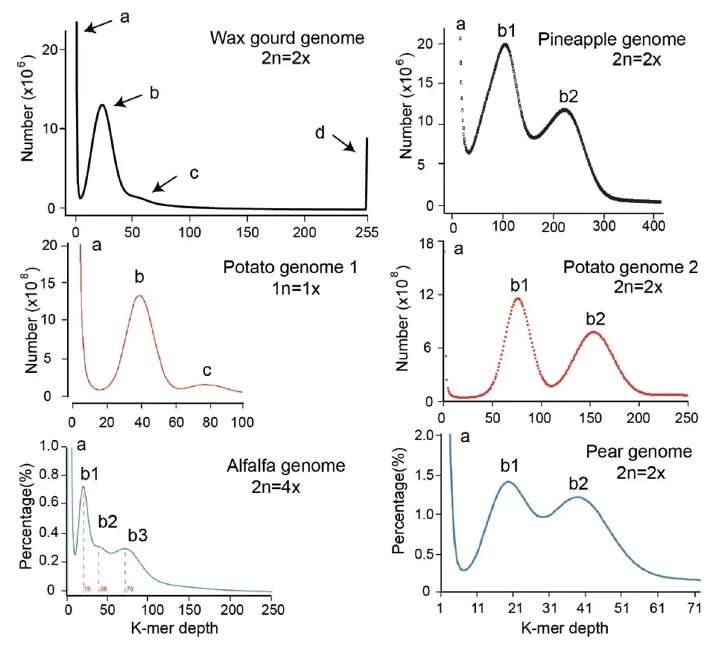 Fig. 3. K-mer distribution curves of Illumina sequencing data of several plant genomes.(Tang Die;, et.al, 2021).
Fig. 3. K-mer distribution curves of Illumina sequencing data of several plant genomes.(Tang Die;, et.al, 2021).
Assembly Strategies for Highly Heterozygous Genomes
Highly heterozygous genomes pose challenges for traditional whole-genome assembly due to significant differences between homologous regions, resulting in numerous branching structures and low contiguity. Employing a phased assembly strategy or extracting haploid data from sequencing datasets can effectively reduce assembly complexity. Modern approaches, such as 10× Genomics technology, utilize long-range information to build long scaffolds, enabling efficient reference genome construction for highly heterozygous genomes. Phased assembly has become a focus in heterozygous species research, providing a foundation for exploring genome diversity in greater depth.
In addition to genome analysis, epigenomic studies offer new insights into gene expression regulation. Epigenetics reveals processes such as gene expression regulation, chromatin remodeling, and gene silencing, all of which play significant roles in plant growth and development. Variant detection helps to identify genetic differences, especially how plants adapt and evolve under varying environmental conditions.
Applications of Genome-Wide Analysis in Plants
1. Crop Improvement and Genetic Modification
Genome-wide analysis provides a solid theoretical foundation and technical support for crop breeding. By deeply analyzing genome data, scientists can identify key genes associated with traits like crop quality, yield, and disease resistance. This information can be used for precision breeding, helping to improve crops' drought tolerance, disease resistance, and nutritional content(Kim, K. D.,et.al, 2020).
2. Identification and Utilization of Disease Resistance Genes
Through genome-wide analysis, researchers can identify genes related to plant disease resistance. These genes play a crucial role in understanding the mechanisms by which plants defend themselves against pathogens. For instance, the identification of Nucleotide Binding Site-Leucine Rich Repeat (NLR) genes has been pivotal in breeding programs aimed at enhancing resistance to diseases such as rust in wheat. The Lr10 gene in wheat is a well-documented example that confers resistance to leaf rust, significantly reducing yield losses due to this pathogen(Tirnaz, S.,et.al,2020).
3. Environmental Adaptation and Stress Tolerance Gene Discovery
Plants must adapt to a variety of environmental stresses such as drought, heat, and salinity during their growth. Genome-wide analysis helps identify genes involved in environmental adaptation and stress tolerance.
4 Metabolic Pathway Analysis and Biosynthesis Research
Genome-wide analysis also aids in uncovering the composition and regulatory mechanisms of plant metabolic pathways. Plants produce a range of secondary metabolites, which are crucial for growth, reproduction, and disease resistance. By analyzing genome data, scientists can identify key enzymes and pathways involved in the biosynthesis of these metabolites, providing a foundation for optimizing plant secondary metabolites or developing plant-based pharmaceuticals.
or instance, studies on the flavonoid biosynthesis pathway in plants like Arabidopsis have led to the identification of key enzymes such as chalcone synthase, which are crucial for producing flavonoids that enhance plant defense against UV radiation and pathogens(Jeffrey ,et.al ,2004).
Bioinformatics Tools and Databases
With the continuous advancement of genomic research, numerous bioinformatics tools and databases have been developed, significantly advancing plant genome analysis. Commonly used tools include GATK, BWA, STAR, and Cufflinks, which facilitate efficient genome assembly, annotation, and analysis. Meanwhile, plant genome databases like Ensembl Plants, TAIR, and PlantGDB provide extensive genomic data, enabling researchers to query and analyze plant genomes.
Conclusion
Genome-wide analysis has become an indispensable tool in plant research. It has not only facilitated crop improvement and environmental adaptation studies but also provided essential technical support for basic plant biology. Moving forward, with advancements in sequencing technologies and computational capabilities, plant genome analysis will provide more comprehensive solutions for precision agriculture, enhancing crop yield, resilience, and nutritional quality. The integration of genomics with other disciplines will open up broader research avenues and further advance the field of plant science.
References:
- Gladman, N., Goodwin, S.et.al. (2023). Era of gapless plant genomes: innovations in sequencing and mapping technologies revolutionize genomics and breeding. Current opinion in biotechnology, 79, 102886. https://doi.org/10.1016/j.copbio.2022.102886
- Li, M., Zhang, D., Gao, Q. et al. Genome structure and evolution of Antirrhinum majus L. Nature Plants 5, 174–183 (2019). https://doi.org/10.1038/s41477-018-0349-9
- Tang Die; Zhou Q. (2021). Advances in plant genome assembly technology. Biotechnology Bulletin. 6,1-12
- Kim, K. D., Kang, Y., & Kim, C. (2020). Application of Genomic Big Data in Plant Breeding:Past, Present, and Future. Plants (Basel, Switzerland), 9(11), 1454. https://doi.org/10.3390/plants9111454
- Tirnaz, S., Bayer, P. E., et.al. (2020). Resistance Gene Analogs in the Brassicaceae: Identification, Characterization, Distribution, and Evolution. Plant physiology, 184(2), 909–922. https://doi.org/10.1104/pp.20.00835
- Jeffrey Chen, Z., Wang, J., et.al. (2004). The development of an Arabidopsis model system for genome-wide analysis of polyploidy effects. Biological journal of the Linnean Society. Linnean Society of London, 82(4), 689–700. https://doi.org/10.1111/j.1095-8312.2004.00351.x

 Sample Submission Guidelines
Sample Submission Guidelines


