
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Epigenetics encompasses a pivotal framework wherein heritable changes occur in the genome without concomitant alterations to the underlying DNA sequence, thereby resulting in observable phenotypic variation. This discipline is widely regarded as a significant branch of genetics. A common misconception posits that epigenetic modifications lack heritability; however, it is well-documented that such modifications can be transmitted across generations.
Within the domain of epigenetics, DNA methylation assumes a central role and is a primary subject of investigation. DNA methylation exerts substantial influence on chromatin architecture, DNA conformation, genomic stability, and the interaction modalities between DNA and associated proteins. Recent advances in the elucidation of DNA methylation have been accompanied by considerable progress in detection methodologies, with methylation array technology emerging as a predominant tool in this arena.
This article endeavors to present a thorough and detailed exploration of DNA methylation and methylation array technologies, thereby providing an exhaustive understanding of these essential facets.
DNA methylation arrays constitute high-throughput analytical instruments designed for the comprehensive evaluation of CpG site methylation across the genome. DNA methylation, acknowledged as one of the most intensively investigated epigenetic modifications, facilitates the covalent bonding of a methyl group to the 5th carbon atom of cytosine within CpG dinucleotides (5'-3' orientation), without modifying the primary DNA sequence. This biochemical event results in the formation of 5-methylcytosine (5mC) and markedly affects gene expression patterns.
DNA methylation is integral to the regulation and reprogramming of gene expression, encompassing a range of critical physiological processes. Such processes include gene silencing, genomic imprinting, X-chromosome inactivation, and the establishment and maintenance of cell-specific expression programs. It is noteworthy that DNA methylation is the most prevalent and common epigenetic modification in mammals, including Homo sapiens and Mus musculus.
These arrays enable the detailed examination of methylation patterns associated with numerous biological processes and pathological conditions, such as carcinogenesis, developmental anomalies, and aging.
DNA methylation arrays utilize microarray technology to measure methylation levels at thousands to millions of CpG sites simultaneously. This is achieved by hybridizing bisulfite-converted DNA to probes on the array. The bisulfite conversion process deaminates unmethylated cytosines to uracil, while methylated cytosines remain unchanged, allowing for differentiation between methylated and unmethylated sites.
DNA methylation arrays offer several advantages over other methods of methylation analysis, such as whole-genome bisulphite sequencing (WGBS) and targeted bisulphite sequencing:
Methylation arrays allow for the high-throughput screening of DNA methylation across the entire genome. This capability enables researchers to examine thousands of CpG sites simultaneously, providing a comprehensive overview of the methylation landscape. For example, Bibikova et al. (2006) demonstrated that methylation arrays could effectively profile DNA methylation in cancer genomes, identifying critical epigenetic alterations associated with tumorigenesis .
Methylation arrays are designed to offer high precision and sensitivity in detecting methylation changes at single-base resolution. This accuracy is crucial for identifying subtle methylation differences that may have significant biological implications. A study by Sandoval et al. (2011) highlighted the ability of methylation arrays to detect early epigenetic changes in colorectal cancer, facilitating early diagnosis and potential intervention .
Compared to whole-genome bisulphite sequencing (WGBS), methylation arrays provide a cost-effective alternative for genome-wide methylation analysis. While WGBS offers comprehensive coverage, it is often prohibitively expensive for large-scale studies. Methylation arrays, on the other hand, deliver substantial data at a fraction of the cost, making them accessible for broader applications. For instance, a cost comparison by Rakyan et al. (2011) emphasized the economic advantages of using methylation arrays for large cohort studies in epigenetics.
One of the significant advantages of methylation arrays is their reproducibility and reliability across different studies and laboratories. The standardized protocols and robust platform design contribute to consistent and comparable results. A meta-analysis by Liu et al. (2014) confirmed the reproducibility of methylation array data, supporting their use in multi-center epigenetic research .
Methylation arrays are versatile tools used in various research applications, from basic science to clinical diagnostics. They are employed to study diverse biological processes, including development, disease progression, and response to treatment. For example, Hirst and Marra (2010) utilized methylation arrays to investigate the epigenetic regulation of gene expression in stem cells, shedding light on the mechanisms underlying cellular differentiation .
Methylation arrays can be integrated with other omics data, such as transcriptomics and proteomics, to provide a holistic view of biological systems. This integration facilitates the correlation of DNA methylation patterns with gene expression and protein activity, enhancing the understanding of complex regulatory networks. The study by Laird (2010) illustrated the power of combining methylation data with gene expression profiles to uncover epigenetic markers in breast cancer.
Illumina methylation microarrays are powerful tools for genome-wide DNA methylation profiling. These arrays leverage Illumina's proven chemistries and high-density bead-based technology to provide single-nucleotide resolution and comprehensive coverage of the methylome.
The progression of Illumina methylation microarrays from the 450K array in 2010 to the 850K array in 2015 and, most recently, the 935K array released in November 2023, reflects substantial advancements in epigenetic research within the domains of human health and disease. DNA methylation analysis has provided critical insights into gene regulatory mechanisms and has been instrumental in the identification and validation of biomarkers. In March 2024, Illumina introduced a novel methylation array, the Methylation Screening Array (MSA), featuring 270,000 CpG sites. This array is designed with a focus on common human traits, prevalent disease phenotypes, environmental exposures, aging, and cell type-specific markers. It is aimed at specialized disease cohorts and large-scale health screening applications, emphasizing emerging functional genomics biomarkers and providing a targeted approach for disease-specific research and extensive health screenings.
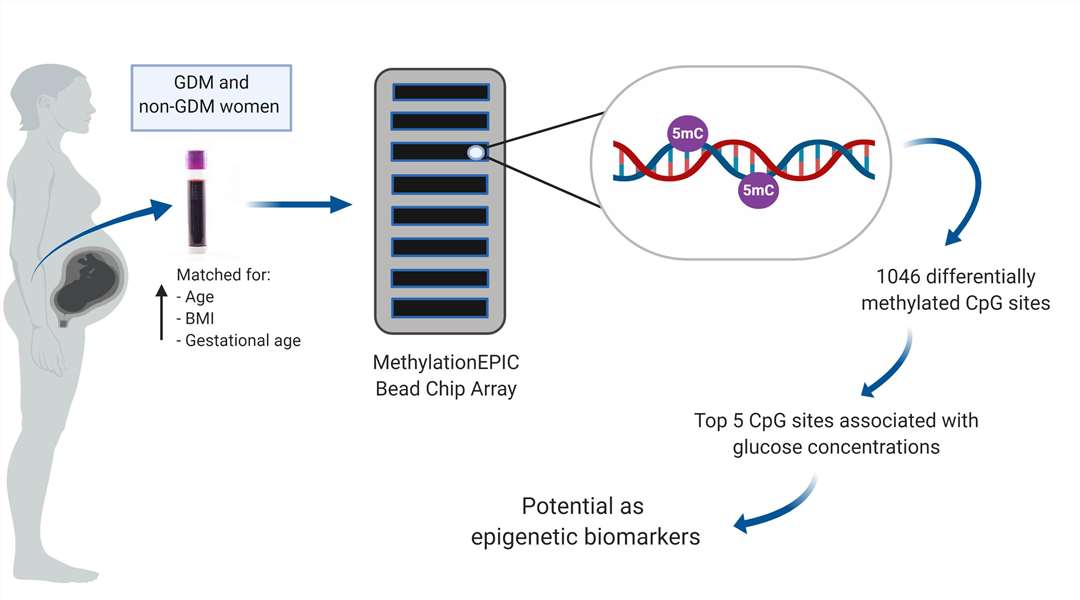 Gestational diabetes mellitus Infinium Methylation EPIC Assay Workflow (Dias, S.et al,. Mellitus. Int. J. Mol. Sci. 2019)
Gestational diabetes mellitus Infinium Methylation EPIC Assay Workflow (Dias, S.et al,. Mellitus. Int. J. Mol. Sci. 2019)
The Infinium Methylation EPIC array, commonly referred to as the 850K methylation array, retains over 90% of the CpG sites from its predecessor, the Infinium Human Methylation450 array. Additionally, it incorporates 350,000 CpG sites in enhancer regions, enabling the detection of over 850,000 methylation sites across the human genome. This array offers a robust solution for studying epigenetic variations by providing quantitative methylation assessments at individual CpG sites from both standard and formalin-fixed paraffin-embedded (FFPE) samples. It is specifically designed for human specimens, reflecting its focused application in human epigenetic research.
Table 1: Coverage of Infinium MethylationEPIC Array across Gene Regions
| Region Type | Number of Sites | Coverage (%) | Average Length (kb) |
| Island | 26,000 | >95% | 6 |
| North shore | 25,000 | >90% | 3.5 |
| South shore | 25,000 | >90% | 3.5 |
| North shelf | 22,000 | >80% | 2 |
| South shelf | 22,000 | >80% | 2 |
The 850K methylation array is employed in epigenome-wide association studies (EWAS) across various human tissue samples. In addition to targeting CpG sites, this array encompasses numerous other valuable loci:
a. CpG sites located outside CpG islands; b. Non-CpG methylation sites (CHH) identified in human stem cells; c. Differentially methylated loci between tumor and normal tissues in various tissue types; d. FANTOM5 enhancers; e. ENCODE open chromatin regions and enhancers; f. DNase hypersensitive sites; g. miRNA promoter regions; h. Over 90% of the markers from the Infinium Human Methylation450 array.
The Infinium Mouse Methylation BeadChip array contains 285K methylation detection sites, enabling precise methylation assessment at the individual CpG site level. This array demonstrates high technical reproducibility, with a parallel reproducibility rate of 98%. It provides comprehensive genomic coverage, including CpG islands, gene transcription start sites, gene body regions, repetitive element regions, enhancer regions, and transcription factor binding sites.
The InfiniumTM MethylationEPIC v2.0 (950K) array represents the latest advancement in Illumina's suite of DNA methylation microarrays. This enhanced version builds upon the robust foundation of the Infinium MethylationEPIC (850K) array, offering expanded coverage and refined assay design to meet the growing demands of epigenetic research.
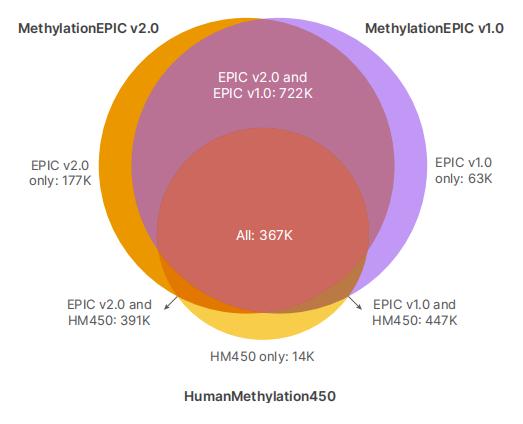 MethylationEPIC v2.0 VS. MethylationEPIC v1.0 ( source for illumina)
MethylationEPIC v2.0 VS. MethylationEPIC v1.0 ( source for illumina)
The Infinium™ MethylationEPIC v2.0 array extends its coverage to over 950,000 CpG sites, thereby significantly enhancing the scope of methylation site analysis in comparison to its predecessor. This enhancement encompasses:
Incorporation of Recent Epigenomic Data: The 950K array integrates recently identified CpG sites derived from contemporary epigenomic studies. This integration ensures that researchers have access to cutting-edge insights and newly identified regions of interest within the genome.
Broad Genomic Representation: The array includes CpG sites distributed across gene bodies, promoters, enhancers, and other regulatory elements, thus facilitating a comprehensive analysis of the methylome. This inclusive representation allows for an extensive interrogation of the genomic landscape.
Diverse Biological Contexts: The expanded coverage addresses various tissues and cell types, enabling the capture of methylation patterns pertinent to a wide range of biological processes and pathological conditions. This diversity ensures the relevance of the methylation data across a multitude of biological and disease contexts.
The InfiniumTM MethylationEPIC v2.0 (950K) array utilizes an optimized combination of Infinium I and Infinium II assay chemistries, enhancing both the depth and sensitivity of methylation detection.
Infinium I Assay Chemistry: This chemistry provides high specificity and sensitivity for CpG sites with dense methylation, ensuring accurate detection in regions with high methylation density.
Infinium II Assay Chemistry: This chemistry is designed for broader coverage, capturing CpG sites with variable methylation patterns. The combination of these two chemistries allows for a balanced and comprehensive methylation profile.
Increased Detection Sensitivity: The refined assay design improves the sensitivity of detection, enabling the identification of subtle methylation changes that may be critical in disease and developmental studies.
The InfiniumTM MethylationEPIC v2.0 (950K) array is designed for high throughput, making it suitable for large-scale studies. Key features contributing to its efficiency include:
Scalability: The array can process a large number of samples simultaneously, facilitating population-scale studies and cohort analyses.
Streamlined Workflow: Similar to other Infinium arrays, the 950K array employs a PCR-free protocol, reducing complexity and minimizing the risk of amplification bias.
Automation Compatibility: The array's workflow is compatible with automated platforms, increasing throughput and reducing hands-on time for researchers.
The augmented coverage and improved design of the Infinium™ MethylationEPIC v2.0 array render it an invaluable instrument for a myriad of applications within the field of epigenetics:
Epigenome-Wide Association Studies (EWAS): The extensive coverage afforded by the 950K array facilitates the identification of differentially methylated regions (DMRs) associated with various diseases and phenotypic traits across diverse populations. This capability is integral to advancing our understanding of epigenetic contributions to complex traits and disease etiologies.
Cancer Research: The array's precision in detecting subtle methylation alterations is of paramount importance in oncological research. Epigenetic modifications frequently play pivotal roles in the processes of tumorigenesis and cancer progression, making high-resolution methylation analysis a critical component of cancer studies.
Developmental Biology: Researchers can leverage the array to investigate methylation patterns throughout developmental stages, thereby elucidating the regulatory mechanisms underlying gene expression and the establishment of cellular identities. This application provides key insights into developmental biology and differentiation processes.
Environmental Epigenetics: The array is also applicable in studying the impact of environmental factors on DNA methylation. This research is essential for understanding gene-environment interactions and their implications on health outcomes, furthering our knowledge of epigenetic responses to environmental stimuli.
The Methylation Screening Array (MSA 270K) by Illumina is a highly specialized microarray designed for targeted methylation analysis. It provides focused coverage of CpG sites relevant to specific research areas, offering a cost-effective and efficient solution for large-scale methylation studies. Its focused coverage of disease-associated CpG sites and regulatory elements makes it an invaluable tool for biomarker discovery, comparative studies, and longitudinal research.
The MSA 270K array features a carefully curated selection of approximately 270,000 CpG sites. These sites have been strategically chosen based on their relevance to various biological and clinical research applications.
Disease-Associated CpG Sites: The array includes CpG sites known to be associated with major diseases, such as cancer, cardiovascular diseases, and neurological disorders. This targeted approach allows researchers to concentrate on regions with established clinical significance.
Regulatory Elements: Coverage extends to CpG sites within promoters, enhancers, and other regulatory elements. This ensures that key regulatory regions are included in the analysis, providing insights into the epigenetic regulation of gene expression.
Tissue-Specific Methylation: The selected CpG sites represent methylation patterns specific to different tissues and cell types, enabling studies that require tissue-specific methylation profiling.
The MSA 270K array is designed to offer a cost-effective alternative to more comprehensive arrays while maintaining high-quality data output.
Reduced Costs: By focusing on a targeted set of CpG sites, the MSA 270K array reduces reagent and processing costs. This makes it an attractive option for large-scale studies and institutions with budget constraints.
Efficient Use of Resources: The array's targeted approach ensures that resources are used efficiently, concentrating on regions most likely to yield meaningful results. This enhances the overall cost-effectiveness of the research.
The MSA 270K array supports high-throughput processing, making it suitable for extensive population studies and large sample cohorts.
Scalable Workflow: The array's workflow is designed for scalability, allowing for the simultaneous processing of numerous samples. This is particularly beneficial for studies involving large populations or multiple time points.
Automation Compatibility: Similar to other Illumina arrays, the MSA 270K is compatible with automated laboratory platforms. This reduces manual labor and increases throughput, ensuring that large datasets can be generated efficiently.
The targeted nature of the MSA 270K array makes it a valuable tool for a variety of epigenetic research applications.
Biomarker Discovery: The array's focus on disease-associated CpG sites facilitates the discovery of methylation biomarkers for early diagnosis, prognosis, and therapeutic response monitoring.
Comparative Studies: Researchers can use the MSA 270K array to compare methylation patterns across different conditions, such as healthy vs. diseased tissues, providing insights into disease mechanisms and potential therapeutic targets.
Longitudinal Studies: The array's cost-effectiveness and high throughput capabilities make it ideal for longitudinal studies that track methylation changes over time, offering insights into dynamic epigenetic modifications.
Despite its targeted focus, the MSA 270K array maintains high standards of data quality and reproducibility.
Robust Assay Design: The array employs Illumina's proven assay chemistries, ensuring reliable and accurate methylation detection.
Reproducible Results: Extensive validation and quality control measures ensure that the data generated by the MSA 270K array are highly reproducible, providing confidence in the research findings.
| Feature | Infinium Human Methylation EPIC (850K) | Infinium Methylation EPIC v2.0 (950K) | Methylation Screening Array (MSA 270K) | Infinium Mouse Methylation BeadChip (285K) |
| Number of Methylation Sites | 850,000 | 950,000 | 270,000 | 285,000 |
| CpG Sites | >90% from 450K array | Enhanced coverage of CpG sites | Focus on common traits and diseases | Focus on CpG and non-CpG sites |
| CpG Islands | Included | Included | Included in broader context | Included |
| Enhancer Regions | FANTOM5 and ENCODE | Enhanced coverage | Targeted coverage | ENCODE |
| Open Chromatin Regions | ENCODE | Enhanced coverage | Included | Covered |
| Non-CpG Methylation Sites | Not included | Not included | Not specifically targeted | CHH sites in stem cells |
| DNase Hypersensitive Sites | Included | Enhanced coverage | Included | Covered |
| miRNA Promoter Regions | Included | Enhanced coverage | Targeted | Included |
| Technical Reproducibility | Not specified | Not specified | Not specified | 98% |
| Species Coverage | Human | Human | Human | Mouse |
| Application Focus | Epigenome-wide association studies | Enhanced resolution for research | Disease phenotype and health screening | Comprehensive genomic coverage |
| Additional Notes | Comprehensive coverage across CpG sites and various regions | Expanded coverage with additional CpG sites | Targeted at emerging functional genomics markers | Covers entire genome, including various regions |
Selecting the appropriate Illumina Methylation Microarray depends on the specific needs of your research and the desired resolution of methylation analysis. Here are the key considerations for choosing between the Infinium MethylationEPIC (850K), Infinium MethylationEPIC v2.0 (950K), and Methylation Screening Array (MSA 270K):
Broad Methylation Studies: For comprehensive genome-wide studies and biomarker discovery, the Infinium MethylationEPIC (850K) array is suitable due to its extensive coverage of over 850,000 CpG sites.
Detailed Methylation Profiling: If your study requires higher resolution and more extensive coverage, the Infinium MethylationEPIC v2.0 (950K) array, with over 950,000 CpG sites, is the best choice. It includes additional CpG sites for a more detailed analysis.
Initial Screening: For targeted methylation studies or initial screening where cost-effectiveness is a priority, the Methylation Screening Array (MSA 270K) with over 270,000 CpG sites is appropriate. It offers focused coverage suitable for specific target studies.
Comprehensive Genome-wide Coverage: Choose the Infinium MethylationEPIC (850K) or Infinium MethylationEPIC v2.0 (950K) arrays for studies requiring broad coverage across the genome. The latter provides an even more extensive and detailed coverage.
Focused Coverage: The MSA 270K is designed for focused coverage, making it ideal for studies that require a cost-effective approach with sufficient resolution for specific targets.
All three arrays (850K, 950K, and 270K) are compatible with various sample types, including FFPE and fresh frozen tissues. This flexibility ensures that you can choose any of these arrays based on other factors without worrying about sample compatibility.
High Reproducibility: All arrays demonstrate high reproducibility (>98% for technical replicates), ensuring reliable results across different experiments.
Analytical Sensitivity: Each array maintains a high analytical sensitivity, with a delta-beta value of 0.2 and a false positive rate of less than 1%. This consistency across arrays means you can select based on coverage and resolution without compromising sensitivity.
All arrays are supported by the GenomeStudio Methylation Module, facilitating integrated analysis. This ensures that data analysis and interpretation are streamlined, regardless of the chosen array.
Budget Constraints: For large-scale screening projects where budget is a significant concern, the MSA 270K offers a cost-effective solution without sacrificing analytical performance.
Detailed Analysis Requirements: For projects where detailed methylation profiling is critical, investing in the Infinium MethylationEPIC v2.0 (950K) may provide the most value due to its extensive coverage.
Infinium MethylationEPIC (850K): Suitable for broad methylation studies and biomarker discovery, providing comprehensive coverage and high-throughput capabilities.
Infinium MethylationEPIC v2.0 (950K): Ideal for detailed methylation profiling and comprehensive analysis with higher resolution.
Methylation Screening Array (MSA 270K): Best for initial methylation screening and specific target studies, offering a balance between cost and coverage.
By evaluating these factors, researchers can choose the most appropriate Illumina Methylation Microarray that aligns with their specific research goals, budget, and desired resolution. CD Genomics offers all three options, providing flexibility and support to meet diverse research needs.
Some Available DNA methylation microarray Services at CD Genomics
| Microarray | Species | Coverage | Platform |
| Infinium MethylationEPIC Array | Human | Detects over 935,000 CpG sites, covering CpG islands, promoters, coding regions, and enhancers comprehensively. | Illumina |
| Infinium Human Methylation 935K | Human | Over 850,000 methylation sites per sample at single-nucleotide resolution | Illumina |
| Human CpG Island Microarray | Human | 27,800 CpG islands covering 21 MB | Agilent |
| Human DNA Methylation Microarrays | Human | 27,627 expanded CpG islands and 5081 UMR regions | Agilent |
| Mouse CpG Island Microarray | Mouse | 15,342 CpG islands | Agilent |
| Infinium Methylation Screening Array 270K | Human | Covers approximately 270,000 methylation sites, with core applications in specific disease cohort research and extensive health screenings. Ensures high-level precision, elevating single array detection throughput by 48 samples, a six-fold increase from the Infinium MethylationEPIC v2.0, achieving higher throughput and lower costs. | Illumina |
Extensive CpG Coverage: The 450K array covers over 450,000 CpG sites, while the EPIC array extends this coverage to over 850,000 sites, encompassing a vast majority of known regulatory regions.
High Sensitivity and Specificity: Infinium technology ensures high sensitivity for detecting methylation changes, even at low levels, and high specificity for accurately distinguishing between methylated and unmethylated cytosines.
Flexible Sample Input: These arrays are compatible with various sample types, including low-input and degraded DNA from FFPE tissues, making them versatile for different research and clinical scenarios.
Comprehensive Annotations: The arrays come with detailed annotations, including gene-centric, CpG island, and regulatory region information, aiding in the interpretation of methylation data in a biological context.
Support for Advanced Bioinformatics: Illumina provides comprehensive bioinformatics support, with tools and pipelines for data processing, normalization, and analysis, facilitating the extraction of meaningful insights from the methylation data.
The workflow for DNA methylation analysis starts with the extraction of DNA from both fresh and preserved tissue samples. For fresh-frozen tissues, standard DNA extraction methods are employed to ensure high-quality DNA. Preserved tissues, such as those fixed in formalin and embedded in paraffin (FFPE), require specialized extraction techniques to recover DNA that may be fragmented and chemically modified. These methods are designed to yield DNA suitable for high-throughput analysis, ensuring the integrity and quality required for accurate methylation profiling.
After extraction, DNA concentration and purity are measured using spectrophotometric techniques and fluorometric assays. Accurate quantification is critical for subsequent steps, while purity assessments help ensure minimal contamination. The integrity of the DNA is often verified through gel electrophoresis, which provides a visual confirmation of DNA size and quality. These quality control measures are essential to guarantee that the DNA is suitable for the methylation array analysis.
The bisulphite conversion of DNA is a pivotal step in methylation analysis. This chemical treatment converts unmethylated cytosines to uracil, while methylated cytosines remain unchanged. The process is essential for differentiating between methylated and unmethylated cytosines, allowing for the accurate detection of DNA methylation patterns. Effective bisulphite conversion is crucial for obtaining reliable data from methylation arrays, as incomplete conversion can lead to inaccurate results.
For samples extracted from preserved tissues, additional steps may be required to restore the bisulphite-converted DNA. This often involves chemical treatments to denature and purify the DNA, improving its quality for hybridization. These restoration procedures enhance the quality of the bisulphite-treated DNA, making it more suitable for subsequent analysis.
The hybridization process involves applying the bisulphite-converted DNA to a microarray containing probes specific to CpG sites. During hybridization, the DNA binds to these probes, and the methylation status is detected based on the binding pattern. This step is crucial for generating the data needed to assess DNA methylation across the genome.
Following hybridization, single-base extension is used to incorporate labeled nucleotides complementary to the target sequences. This step enhances the sensitivity of the assay, allowing for precise measurement of methylation levels at individual CpG sites. The intensity of the fluorescence signal correlates with the degree of methylation, providing quantitative data on DNA methylation patterns.
The raw data from the methylation array are first normalized to account for technical variability and background noise. Various quality control measures are implemented, such as excluding probes with high detection p-values or those on sex chromosomes. These steps ensure that the data are reliable and accurate for downstream analysis.
Advanced computational tools are used to analyze the methylation data, including principal component analysis (PCA) and clustering methods. These analyses help identify patterns and associations within the data, providing insights into the biological implications of DNA methylation changes.
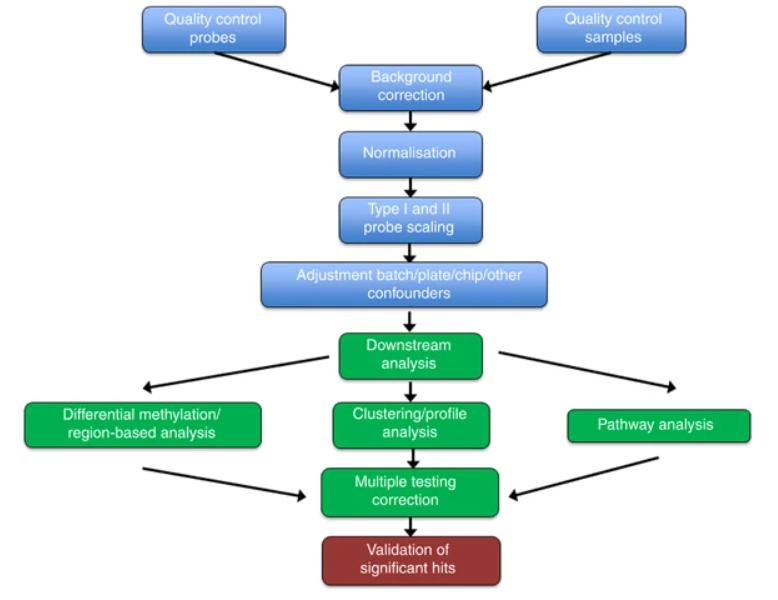 Methylation array data processing and analysis pipeline. (C.S. Wilhelm-Benartzi, et al,. British Journal of Cancer 2013)
Methylation array data processing and analysis pipeline. (C.S. Wilhelm-Benartzi, et al,. British Journal of Cancer 2013)
Illumina methylation microarrays are powerful tools for investigating the epigenetic landscape across the genome. These arrays are widely used in various fields of biomedical research due to their high resolution, cost-effectiveness, and ability to measure methylation levels at hundreds of thousands of CpG sites. The applications of Illumina methylation microarrays span cancer research, developmental biology, neuroscience, environmental epigenetics, and personalized medicine.
Illumina methylation microarrays have significantly advanced cancer research by enabling the identification of tumor-specific methylation patterns. Aberrant DNA methylation is a hallmark of cancer, often involving hypermethylation of tumor suppressor genes and hypomethylation of oncogenes. For example, a study by Sandoval et al. (2011) used the Illumina HumanMethylation27 and HumanMethylation450 BeadChips to profile methylation in chronic lymphocytic leukemia (CLL) samples, identifying distinct methylation signatures associated with disease subtypes and prognosis . Another study by Aryee et al. (2013) utilized the HumanMethylation450 array to map the methylation landscape of colorectal cancer, revealing key epigenetic alterations driving tumorigenesis.
Methylation microarrays are instrumental in studying the dynamic changes in DNA methylation during development. These changes are crucial for proper gene regulation and tissue differentiation. Laurent et al. (2010) employed the Illumina HumanMethylation27 BeadChip to investigate the methylation patterns in human embryonic stem cells (hESCs) and their differentiated progeny, uncovering stage-specific methylation changes that regulate pluripotency and differentiation . Similarly, Lee et al. (2015) used the HumanMethylation450 array to examine methylation dynamics during early embryonic development, providing insights into the epigenetic reprogramming events that occur post-fertilization.
In neuroscience, Illumina methylation microarrays facilitate the study of epigenetic mechanisms underlying neurological disorders. For instance, Lunnon et al. (2014) utilized the HumanMethylation450 array to identify differential methylation patterns in Alzheimer's disease (AD) brains, highlighting genes involved in immune response and neuronal function as potential contributors to AD pathology . Another study by Numata et al. (2012) used the same array to explore methylation changes in schizophrenia, identifying numerous differentially methylated regions (DMRs) associated with the disorder.
Environmental determinants, including dietary intake, exposure to toxins, and psychological stress, have the capacity to modulate DNA methylation patterns. These modifications bear significant implications for health outcomes and disease susceptibility. Illumina methylation microarrays serve as critical tools for the evaluation of such epigenetic alterations. An illustrative example is provided by an EWAS conducted by Breitling et al. (2011), wherein the HumanMethylation450 array was utilized to examine the influence of smoking on DNA methylation. This study identified multiple CpG sites where methylation status exhibited correlation with smoking behavior. Similarly, Rusiecki et al. (2010) employed the HumanMethylation27 BeadChip for the evaluation of arsenic exposure effects on methylation patterns, leading to the identification of potential biomarkers indicative of arsenic-related pathologies.
The integration of methylation datasets with other omics data holds significant potential for the advancement of personalized medicine. Profiling individual methylation patterns enables the development of bespoke therapeutic strategies. For instance, research conducted by The Cancer Genome Atlas (TCGA) consortium employed Illumina methylation arrays to categorize glioblastoma multiforme (GBM) patients into subgroups characterized by distinct epigenetic and genetic profiles, thereby facilitating personalized treatment approaches. Additionally, methylation biomarkers identified through these arrays can serve as valuable tools in early disease detection, prognosis, and monitoring of treatment responses.
References:


