
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

Copy Number Variation (CNV) is a type of genomic structural variation that encompasses alterations in the number of copies of a particular DNA segment. These variations are broadly categorized into two levels based on their size: microscopic and submicroscopic.
Microscopic genomic structural variations are observable under a microscope and include chromosomal aberrations such as aneuploidy, deletions, insertions, inversions, translocations, and fragile site disruptions. CNV manifests in various forms within the genome, including simultaneous deletion of copies on both homologous chromosomes, deletion on one homologous chromosome with the other remaining normal, and duplication of copies on one homologous chromosome while the other remains normal.
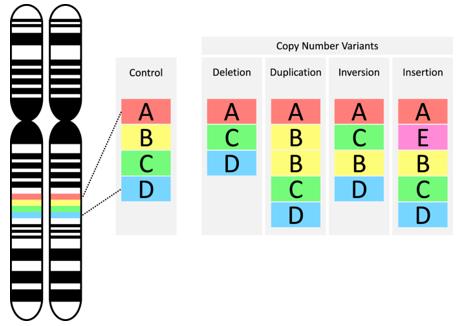 Types of copy number variants (CNVs). (Mollon et al., 2023)
Types of copy number variants (CNVs). (Mollon et al., 2023)
On the other hand, submicroscopic genomic structural variations occur at the DNA fragment level within the 1Kb-3Mb range. These variations include deletions, insertions, duplications, rearrangements, inversions, and changes in DNA copy number, collectively referred to as CNV.
Initially discovered in patients' genomes, CNVs have been found to be prevalent in normal human populations as well, suggesting a spectrum of clinical significance ranging from benign to pathogenic or unknown. The precise mechanism behind CNV formation remains unclear, but potential mechanisms include non-allelic homologous recombination (NAHR) and non-homologous end joining (NHEJ).
Cutting-edge technologies, such as high-throughput sequencing and long-read sequencing, employed by CD Genomics, facilitate the detection of CNV and genotyping. This advanced sequencing approach allows for comprehensive and efficient examination of genetic material, providing valuable insights into the molecular landscape and potential biomarkers associated with various conditions.
Determining the copy number of target fragments through read depth sequencing stands as a powerful method capable of simultaneously detecting multi-gene CNVs and other biomarkers. However, the efficacy of this approach is influenced by a myriad of factors, including panel design, probe GC content, tumor content, and contamination levels, all of which impact the algorithm model's accuracy.
Short-read and long-read sequencing offer a comprehensive view of genomic alterations, allowing researchers to discern copy number variations with precision. By analyzing the depth of reads across target fragments, researchers can unveil alterations in copy number, shedding light on the genomic landscape of interest.
Despite its potential, the effectiveness of sequencing hinges on careful consideration of various factors. Panel design plays a crucial role in determining the regions of interest and ensuring comprehensive coverage across the genome. Similarly, probe GC content influences the efficiency of target capture and sequencing, affecting the accuracy of copy number determination.
Tumors are intricate manifestations of genomic anomalies, arising from a cascade of mutations within the somatic cells. Among these mutations, Copy Number Variations (CNVs) in tumor samples stand out as pivotal players. In normal somatic cells, the genome is diploid; however, in tumor cells, certain genomic regions undergo copy number amplifications or deletions, altering the original genomic landscape within a size range of approximately 50bp to 1Mb.
Deletion mutations in some tumors trigger the activation of proto-oncogenes, leading to oncogene inactivation, as exemplified by RB1, P16, PTEN, and others. Conversely, amplification mutations induce the activation of proto-oncogenes and oncogenes, such as MYC, HER2, EGFR, respectively. These genes intricately participate in various signaling pathways, pivotal in the development and regulation of cellular processes. They wield significant influence over cell growth, proliferation, metastasis, and recurrence.
The detection of tumor-specific CNVs not only offers insights into the molecular underpinnings of tumorigenesis but also accelerates the discovery of novel tumor proto-oncogenes and oncogenes. This knowledge becomes invaluable in the quest for effective therapeutic interventions against tumors. Such studies pave the way for targeted therapy in tumor patients, enabling clinicians to tailor personalized treatments based on individual copy number variations. For instance, drugs like trastuzumab and patozumab exhibit enhanced efficacy in metastatic breast cancer cases characterized by overexpression of the HER2 gene.
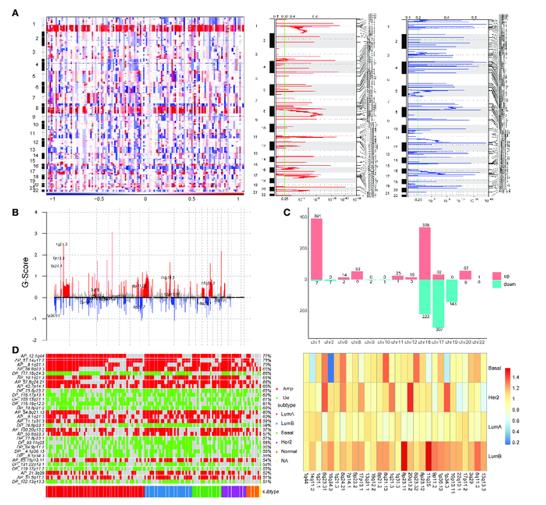 Identification of copy number variation-driven enhancers in breast cancer. (Zhao et al., 2022)
Identification of copy number variation-driven enhancers in breast cancer. (Zhao et al., 2022)
Performing Copy Number Variation (CNV) analysis is a multi-step process, spanning from data preparation to the identification and annotation of CNVs. Below is a comprehensive guide outlining the standard procedure for CNV analysis, utilizing the fq.gz file provided by the sequencing company:
Begin by uncompressing the fq.gz file to obtain raw sequencing data in FASTQ format.
Assess the quality of the sequencing data using quality control tools such as FastQC. Evaluate parameters including base quality scores, sequence quality distribution, and GC content to ensure data integrity.
Align sequencing reads to a reference genome utilizing alignment tools such as BWA or Bowtie.
Process alignment results using tools like SAMtools for format conversion (SAM to BAM), sorting, and de-duplication to streamline downstream analysis.
Calculate read segment coverage for each genomic region based on alignment results.
Utilize tools like BEDTools to generate coverage files for precise characterization of genomic regions.
Employ CNV detection tools such as CNVnator, DELLY, or LUMPY to analyze coverage data.
Identify copy number variations by leveraging read segment coverage, pairing information, and/or split reads.
Filter CNV results based on predefined criteria such as CNV quality, size, and frequency to ensure accuracy.
Employ functional annotation tools like ANNOVAR or VEP to annotate detected CNVs, providing insights into their biological significance.
The UCSC Genome Browser stands as a cornerstone in copy number variant analysis, offering indispensable tools such as the Genome Browser and LiftOver function.
The UCSC Genome Browser serves as a versatile virtual microscope, facilitating seamless navigation through genomic data with interactive graphical displays. Its user-friendly interface streamlines the exploration of genomic landscapes, making data retrieval faster, more accessible, and reliable. By amalgamating a vast array of genome annotation data, this browser empowers researchers to delve deep into the intricacies of the human chromosome genome, down to the minutiae of individual nucleotides. As depicted below, users can input their query information into the designated window, with the annotation display window presenting the findings in an intuitive graphical format.
The DECIPHER database stands as a cornerstone in the realm of bioinformatics, particularly in molecular genetics. It serves as an invaluable resource for researchers seeking comprehensive information on genetic diseases, encompassing mutation loci, clinical phenotypes, and more. Currently housing data from 44,153 patients, DECIPHER offers a rich repository of genetic insights.
Users can easily navigate the database to explore a myriad of genetic disease information, including 65 microdeletion and microduplication syndromes linked to developmental disorders, along with 786 gene disorders meticulously documented in GeneReviews. Each entry provides a detailed description of the disorder, fragment size, literature references, and comprehensive information about the associated genes, variants, and phenotypes.
Querying for Basic Disease Information within Copy Number Variant Segments
Researchers can utilize the DECIPHER database to swiftly query basic disease information within copy number variant segments, such as CNV Syndromes and GeneReviews. The database facilitates efficient retrieval of pertinent data, aiding in the elucidation of genetic disorders and their underlying molecular mechanisms.
Copy Number Variant Number of Protein-Coding Genes within a Fragment Query
The third section of the CNV Scoring Tool in the new ACMG Guidelines relies on the number of protein-coding genes within a copy number variant segment to assign different scores. DECIPHER database offers a seamless platform for querying this crucial information, empowering researchers to make informed decisions. By default, DECIPHER operates on the GRGh38 genome version, with provisions for conversion if the evaluated fragments utilize a different genome version. Caution is advised when evaluating segments containing gene clusters or families. In cases where the clinical significance of a gene family is unclear, each family can be considered as one gene. However, genes with known clinical relevance or clear disease associations should be counted separately, ensuring accuracy in genetic analysis and interpretation.
ClinGen stands as a pivotal resource, generously funded by the National Institutes of Health (NIH), dedicated to curating comprehensive insights into the clinical relevance of genes, variants, and diseases, with a keen focus on advancing precision medicine research. In our pursuit of understanding copy number variations (CNVs), two indispensable tools from ClinGen emerge: the ClinGen-Dosage Sensitivity and the ClinGen CNV Pathogenicity Calculator.
The utilization of ClinGen-Dosage Sensitivity is paramount in CNV analysis, particularly in assessing whether CNVs overlap with genes or regions unequivocally or predicted to exhibit single underdose effects (triple dose-sensitive effects), or conversely, those deemed unequivocally benign. This critical step forms the second part of the CNV scoring tool outlined in the new ACMG guidelines, guiding researchers in discerning the clinical significance of identified CNVs.
ClinGen's robust infrastructure empowers researchers with the necessary tools to navigate the intricate landscape of CNV analysis with precision and confidence. By leveraging ClinGen-Dosage Sensitivity, researchers gain access to curated data essential for making informed decisions regarding the clinical implications of CNVs.
References:


