Complete nucleotide bases in a sample are determined by a sequencer where a short sequence or read for each fragment in the library is generated. Modern sequencing technologies generate huge amounts of sequence reads in just one experiment, but all of them have technical limitations. Next-generation sequencing can be affected by factors during the library preparation and sequencing processes, which can negatively affect the quality of the raw data. Each sequencing procedure will produce different amounts of errors, such as incorrect nucleotides being read and deviation from optimal library fragment sizes. For example, metagenomics deal with large numbers of libraries from gut microbial communities; hence, an error would greatly affect the analysis of protein function and metabolic pathways of specific genes. For meaningful downstream applications, sequence quality control seeks to determine and eliminate errors. This first step helps researchers save time, effort, and resources.
The procedure for quality control protocol should be performed at varying time points of data processing including the correct interpretation of results to ensure a meaningful study. Raw sequencing data quality and depth, read duplication rates, and alignment quality is some of the characteristics that should be evaluated in order to increase the accuracy of the outcome. Quick identification of poor-quality samples relies on controlling the quality of raw data. This means taking extra effort from the beginning to save time for later analysis. Alignment quality is crucial for identifying poor-quality samples that passed the raw data quality control checks and also for successful variant detection. Performing quality control on single nucleotide polymorphisms (SNP) reads identify bad samples and decreases the number of false-positive SNP calls, for the majority of exome sequencing studies.
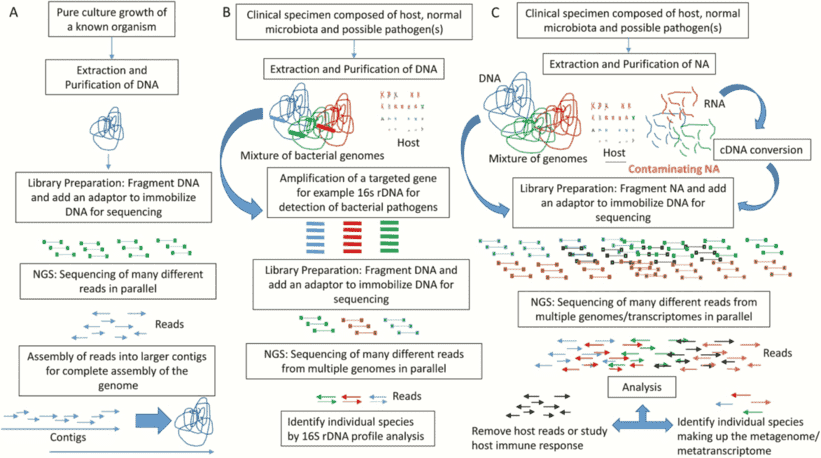
Quality control of NGS data, for example, can be hard for different laboratories with varying resources such as lack of expertise to perform the procedures. Therefore, as shown in Figure 1, standardization and simplification of NGS workflows involving quality control is a central requirement. Each checkpoint corresponds to a cautious examination of the environment, protocol, result, and reagents.
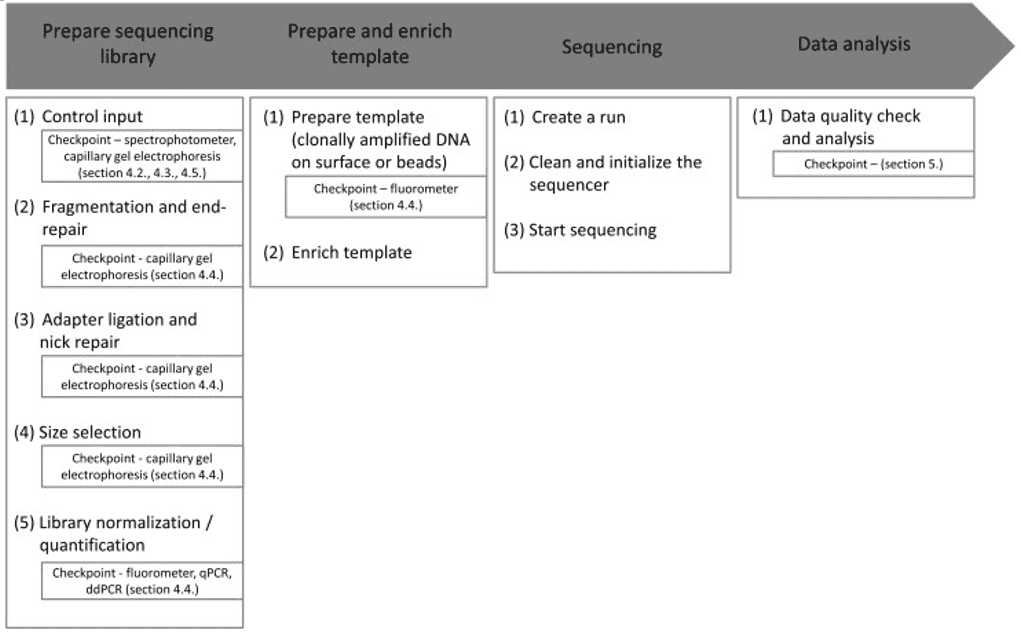 Figure 1. Overview of the general NGS workflow with checkpoints for QC (Endrullat, 2016).
Figure 1. Overview of the general NGS workflow with checkpoints for QC (Endrullat, 2016).
There are two different devices almost exclusively used for QC in sequencing projects. The first is the capillary gel electrophoresis, which studies for investigation of fragment size distribution as well as final library quality assessment. The second most commonly used is the fluorometer for fluorometric quantitation. For accurate quantification of DNA/RNA at certain workflow steps and for determining the final library quantity, systems such as quantitative PCR (qPCR) or digital droplet PCR (ddPCR) are recommended.
Sequence assembly and gene expression studies, which are common in metagenomics, require an excellent quality of data. NGS techniques support in-depth analyses of samples; however, they may still introduce sources of errors and bias. Downplaying these sources or conducting poor quality control can lead to serious issues such as confusing analyzed results and sample-contamination; this could weaken any claims and increase the certainty of the biological outcome.
References
- Endrullat, C., Glökler, J., Franke, P., Frohme, M. Standardization and quality management in next-generation sequencing. Applied & Translational Genomics, 2016, 10, 2–9.
- Trivedi, U. H., Cézard, T., Bridgett, S., et al. Quality control of next-generation sequencing data without a reference. Frontiers in Genetics, 2014, 5.