
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
The skin, measuring approximately 1.8 square meters and regarded as the biggest organ in the human body, is the home of the skin microbiome for millions of bacteria, archaea, fungi, viruses, and small arthropods. It behaves like a physical shield that inhibits foreign pathogens from invading and offers the commensal microbiota with habitat. In addition to being vital in guarding the body against intruding pathogens, skin microorganisms are also essential in educating our immune system and in dissolving natural human products. Owing to its plentiful layers, invagination, and specialized niches, skin can facilitate a wide scope of microorganisms.
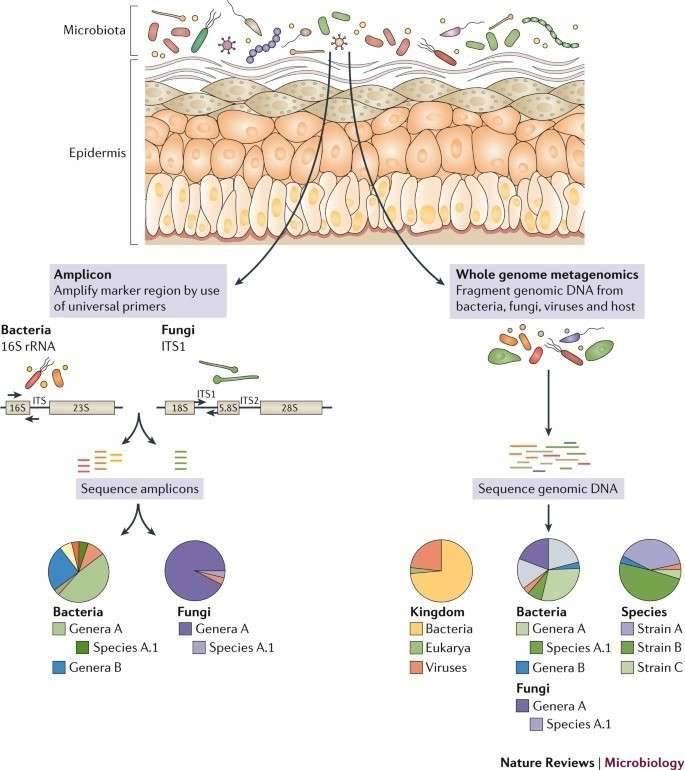 Figure 1. The human skin microbiome. (Byrd, 2018)
Figure 1. The human skin microbiome. (Byrd, 2018)
Many variables, such as host genetic and environmental factors, impact the variability and allocation of skin microorganisms. Even the tough physical status of especially acidic, desiccated, and nutrient-poor skins affects the proportion of the skin microorganism. The following are other established variables: (1) host physiology, (2) the immune system, (3) lifestyle, and (4) the host’s pathobiology or health status.
Various skin microbiome studies intend to recognize the skin by (1) illustrating the mechanisms associated with the coevolution of host organisms and their varied compilation of microorganisms, (2) classifying the immune system in regard to these organisms, (3) diagnosing diseases featuring pathogenic microorganisms, and (4) discovering the etiologies of various skin conditions.
A popular method is based on amplification, sequencing, and assessment of the prokaryotic 16S ribosomal RNA (rRNA) gene to define cutaneous microbial diversity. In several experiments of skin microbial flora and their connection with health and illness, this method has been used. Full-length 16S rRNA gene sequences (∼1500 kb) produced by Sanger sequencing methods were used in preliminary research. Next-generation sequencing frameworks create shorter read lengths that enable significantly greater sequencing depth at a fraction of the price, making it inefficient to sequence the full-length gene. One or more hypervariable areas, or 16S tags, are therefore picked as a proxy for the full-length gene for sequencing. No single hypervariable area can differentiate between all bacteria, and the amplification productivity of various kinds of bacteria can be differentially affected by primary biases. Even so, for seizing the differences and structure of different ecosystems, particular regions may be ideal.
More lately, for both taxonomic and functional annotation of skin microbial populations, whole metagenomic shotgun (WMS) sequencing has been utilized. This strategy decreases the bias of amplification, encapsulates multi-kingdom populations, and enables strain-level assessment. Although data-rich, WMS datasets are more costly to produce and involve higher computational understanding and assets to store, process, and evaluate. Even though gene concentration can be collected from WMS data to give an overview of the bacterial population's operational procedures, bioinformatic methods now arise to foretell functional material from 16S tag sequences and may be preferable in some instances to WMS sequencing for classification of the microbial community.
Use a soft cotton tip swab to wipe the skin to acquire free and attached microbes to acquire skin specimens for testing of the skin microbiome. In terms of moisture (sweat) and sebum, sample skin areas have varied microenvironments: sebaceous (retroauricular crease, occiput, forehead), moist (toe web, umbilicus), and intermittently moist (antecubital fossa, palm). The specimen swabs may be housed for DNA isolation at −20 °C or −80 °C until testing.
References
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to info@cd-genomics.com for inquiries.
Please fill out the form below: ×


Follow us on: