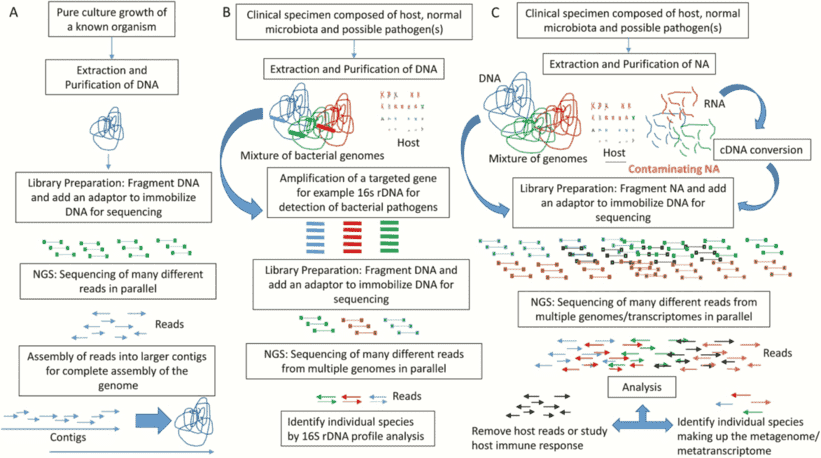
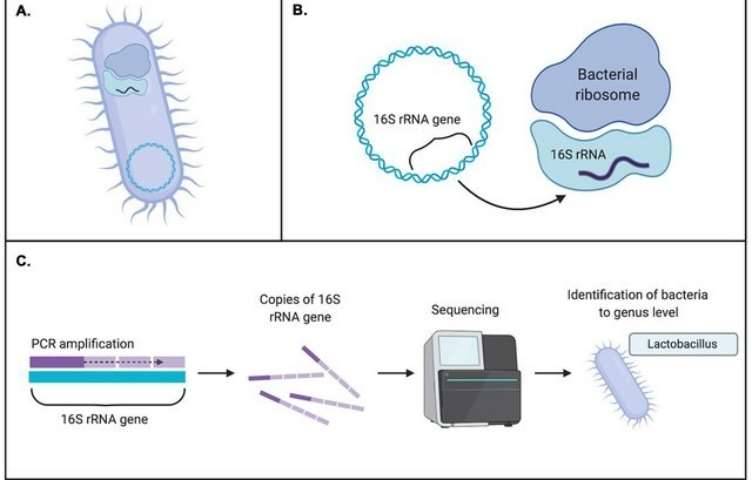
Introduction to Comparative Bacterial Genomics
Comparative genomics is a branch of biology that compares the genome sequences of different species, from microorganisms to human. Researchers can learn what distinguishes different life forms at the molecular level by comparing the sequences of genomes from different organisms. Comparative genomics is also helpful for studying organism evolution because it helps in the discovery of genes that are conserved or shared across species, as well as genes that give each organism its own unique characteristics.
Microbial whole genome sequencing is now fast and inexpensive enough to be considered a tool for bacterial research. This work is done by a diverse group of people who are interested in a wide range of topics related to bacterial genetics and evolution, including researchers, public health practitioners, and clinicians. Clinical isolates, as well as laboratory strains and mutants, are studied, as are outbreak investigations and the evolution and spread of drug resistance. Many labs can now generate bacterial genome sequences in-house in a matter of hours or days using benchtop sequencers like the Illumina MiSeq, Ion Torrent PGM, or Roche 454 FLX Junior.
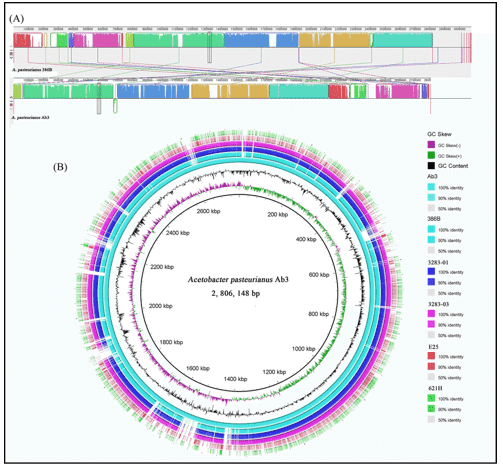 Figure 1. An example of comparative bacterial genome analysis. (Xia, 2016)
Figure 1. An example of comparative bacterial genome analysis. (Xia, 2016)
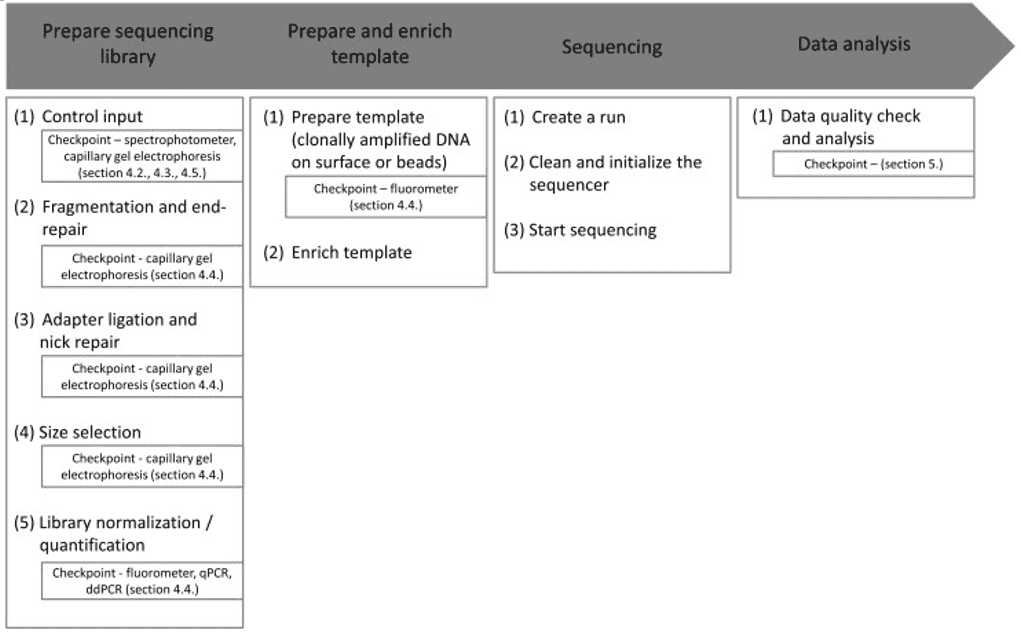
Comparative Bacterial Genome Analysis using Next-Generation Sequence Data: Workflow
Assembly, contig ordering, annotation, genome comparison, and typing are the five logical segments of the workflow.
Genome assembly
The procedure of combining overlapping sequence reads into contiguous sequences (contigs) without using a reference genome as a guide is known as de novo assembly. Short-read sequence assemblers that use de Bruijn graphs to generate an assembly are typically the most efficient. The open-source program Velvet is one of the first and most widely utilized de Bruijn graph assemblers. Velvet stays one of the most-used (and cited) assemblers for bacterial genomes, particularly fits to Illumina sequence reads, with further development to enhance the resolution of repeats and scaffolding using paired-end and longer reads.
Ordering and viewing assembled contigs
The next step is to arrange the contigs against a suitable reference genome after they have been assembled from the sequencing reads. The best reference to use is usually the most closely related bacterium with a 'completed' genome, but finding the best reference, as in the case of E. coli O104:H4, may require trial and error. Contig ordering can be done with command-line tools like MUMmer, which can be made easier with a wrapper program like ABACAS.
Genome annotation
The next step is to annotate the draft genome once the ordered set of contigs has been obtained. Annotation is the process of locating and identifying genes, as well as ribosomal and transfer RNAs encoded in the genome. Uploading a genome assembly to an automated web-based tool like RAST is the most straightforward way to annotate a bacterial genome. Many command-line annotation tools are also available. These include de novo gene discovery methods like Prokka and DIYA, as well as programs like RATT and BG-7 that transfer annotation directly from closely related genomes.
Applications of Comparative Genomics
In the fields of molecular medicine and molecular evolution, comparative genomics has a wide range of applications. The identification of drug targets for many infectious diseases is the most important application of comparative genomics in molecular medicine. The use of comparative genomics can aid in the selection of model organisms. Comparative genomics aids in the clustering of regulatory sites, which can aid in the identification of previously unknown regulatory regions in other genomes.
References
- Merino N, Zhang S, Tomita M, Suzuki H. Comparative genomics of Bacteria commonly identified in the built environment. BMC genomics. 2019 Dec;20(1).
- Yu J, Blom J, Glaeser SP, et al. A review of bioinformatics platforms for comparative genomics. Recent developments of the EDGAR 2.0 platform and its utility for taxonomic and phylogenetic studies. Journal of biotechnology. 2017 Nov 10;261.
- Xia K, Li Y, Sun J, Liang X. Comparative Genomics of Acetobacterpasteurianus Ab3, an acetic acid Producing strain isolated from Chinese traditional rice vinegar meiguichu. PloS one. 2016 Sep 9;11(9).
- Edwards DJ, Holt KE. Beginner's guide to comparative bacterial genome analysis using next-generation sequence data. Microbial informatics and experimentation. 2013 Dec;3(1).