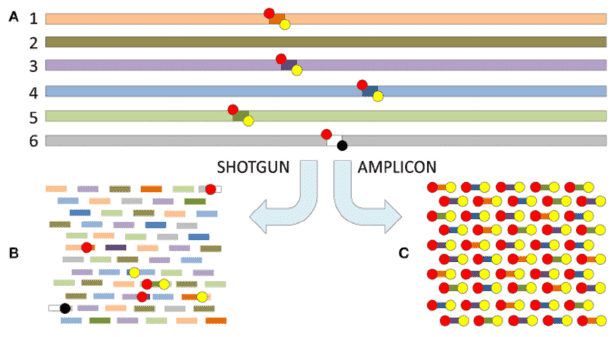
In next-generation sequencing (NGS) technologies, the two predominant library preparation methods—capture-based and amplicon-based strategies—each present unique advantages and limitations. This article aims to elucidate the fundamental distinctions between these approaches, with particular emphasis on their differential tolerances to probe and primer mismatches.
Mismatch Tolerance and its Implications for Library Preparation
Mismatch tolerance, as defined in previous studies (doi:10.1128/mBio.01491-15), refers to the ability of probes to bind to target regions without requiring perfect sequence alignment. It has been demonstrated that a sequence similarity of approximately 75% is generally sufficient for successful probe capture, with some studies suggesting that 70% similarity may suffice in certain contexts. In contrast, primer binding—especially at the 3' end of the primer—requires a higher degree of specificity, with a perfect match being essential for efficient amplification. This stringent requirement for primer sequence fidelity underpins technologies such as ARMS-PCR, which is used for detecting single nucleotide polymorphisms (SNPs).
The divergent mismatch tolerance between these two strategies has significant implications for the selection of reference genomes. Capture-based methods allow for a higher degree of mismatch between the probe and target sequences, enabling the use of reference genomes from species A to design probes for closely related species B. For instance, in phylogenetic studies, probes designed from the reference genomes of several species can be used for species capture sequencing across a broader taxonomic group, facilitating the construction of phylogenetic trees.
Conversely, amplicon-based methods necessitate precise primer matching, particularly for sequences derived from non-target species. The presence of even a few mismatched bases can result in amplification failure. Although transcriptome sequences from the target species are typically used, amplicons that span intronic regions are generally unsuitable for this approach.
The influence of reference genome selection on probe and primer design is summarized in the following table:
| Reference Genome Status |
Liquid Phase Capture |
Multiplex PCR |
Example |
| Complete reference genome available |
√ |
√ |
Rat, Mouse |
| Incomplete reference genome |
√ |
√ |
Potato |
| No reference genome available for target species, but available for a closely related species |
√ |
× |
Ferret |
| No reference genome for target or related species |
× |
× |
Most invertebrates |
| Transcriptomic data available |
√ |
√ |
- |
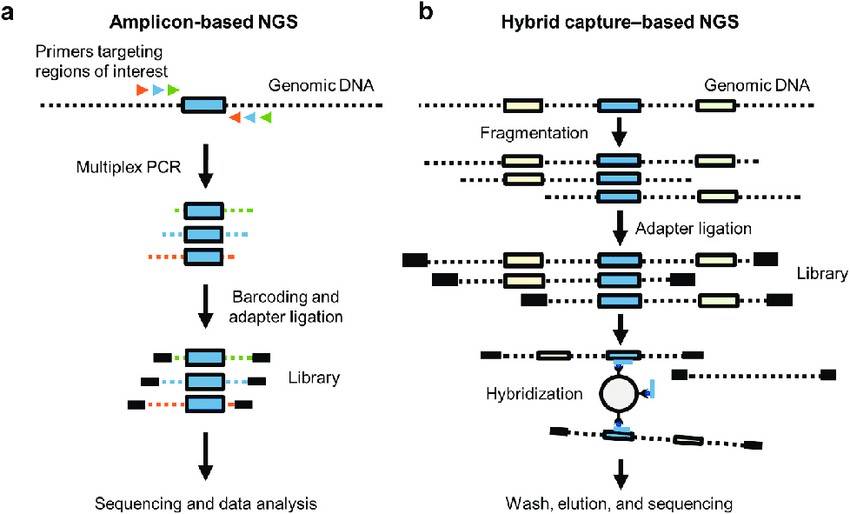 Overview of the amplicon-based and hybrid capture-based NGS methods. (Janakiraman Subramanian et al,.2021)
Overview of the amplicon-based and hybrid capture-based NGS methods. (Janakiraman Subramanian et al,.2021)
Workflow Considerations: Capture vs. Amplicon Methods
A common misconception suggests that multiplex PCR follows a "capture-then-library" workflow, whereas probe capture methods follow a "library-then-capture" sequence. This assertion, however, is not entirely accurate. In fact, there exists an alternative workflow where probes are first used to capture target sequences, after which the captured single-stranded DNA is subjected to library preparation. The single-stranded library preparation method utilized in SWIFT technology is an example of this workflow. It is particularly advantageous when working with cell-free DNA (cfDNA) for methylation capture, where it proves to be an ideal solution.
Furthermore, the view that amplicon-based methods are limited to detecting known mutations such as SNPs and insertions/deletions (InDels) is overly simplistic. While amplicon-based methods are indeed well-suited for detecting hotspots, they are not restricted to this purpose. A more accurate description would be that amplicon-based methods are suitable for detecting point mutations, such as SNPs and InDels. The success of this approach is closely linked to the length of the amplicons; for example, amplicons ranging from 160 to 260 base pairs are suitable for direct NGS analysis. Moreover, certain long PCR techniques have the potential to detect copy number variations (CNVs).
While amplicon-based methods do have limitations in detecting CNVs and structural variants (SVs), these limitations should not be viewed as inherent to the approach. With increased PCR cycle numbers, the inherent instability or preference of PCR amplification may obscure the original CNV data, leading to inaccurate detection. Additionally, SV detection is constrained by the availability of known fusion events. In cases where only one fusion partner's sequence is known, and the other is entirely unknown, primer design becomes challenging. However, for well-characterized fusion events, the design of appropriate primers is straightforward when both ends of the fusion sequence are known. Techniques such as anchored PCR (doi:10.1038/nm.3729) can be employed to detect positional fusions, contributing to the study of SVs, including fusions and viral integrations.
When it comes to detecting point mutations in the coding regions (CDS) of three genes, it is possible to design multiple amplicons for amplification and sequencing. This approach is not limited to detecting hotspots and can also uncover previously unknown mutations, provided they are amplified and sequenced.
Time Considerations in Library Preparation
It is well-established that capture-based methods typically require longer preparation times compared to amplicon-based approaches. However, the time required for capture-based methods is not fixed at 1-2 days. By employing transposase-based library preparation or enzyme digestion methods combined with rapid hybridization techniques (2-hour protocols), library preparation can be completed within 8-12 hours. Nonetheless, the 2-hour rapid hybridization protocol does present certain challenges, particularly with incomplete hybridization, which can lead to reduced product concentration and decreased uniformity under identical post-PCR conditions. Nevertheless, data indicate that the specificity of capture is not significantly compromised under these conditions.
Key Factors to Consider
Starting DNA Quantity: Capture-based methods typically require a higher starting DNA quantity due to the need for DNA fragmentation, which often results in some loss. The greatest loss occurs with ultrasonication, followed by enzyme digestion, with transposase-based methods resulting in the least DNA loss. Conversely, PCR amplification is highly sensitive and can be performed even with minimal template DNA. For example, research on the sequencing of SARS-CoV-2 genomes directly from clinical samples (Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples) indicates that capture methods are more suitable for samples with cycle threshold (Ct) values below 30, whereas PCR amplification can detect variants in samples with Ct values as high as 35.
DNA Integrity: Capture-based methods, which rely on adapter ligation for library construction, can tolerate shorter DNA fragment lengths compared to amplicon-based methods. For example, in ancient DNA studies where DNA fragments are often reduced to lengths of 40-60 base pairs, designing sufficiently short primers to amplify these fragments is challenging. This issue is particularly pertinent in the case of cfDNA amplicon panels, where amplification is difficult. By contrast, capture-based methods do not face this challenge.
Methylation Capture: Currently, second-generation sequencing methods for methylation analysis primarily rely on bisulfite treatment or antibody enrichment. An established principle is that PCR amplification must not occur prior to bisulfite treatment or antibody-based enrichment. Although amplicon-based methods can be used to detect methylation, their applicability is limited to samples with sufficiently abundant gDNA. In contrast, capture-based methods can effectively process cfDNA, a crucial advantage for early cancer screening applications.
Summary of Comparison of Capture-Seq and Amplicon-Seq Library Preparation Methods
| Aspect |
Capture-Seq |
Amplicon-Seq |
| Mismatch Tolerance |
Allows ~70-75% sequence similarity |
Requires a perfect match at 3' end of primer |
| Reference Genome Use |
Utilizes reference genomes from closely related species |
Requires precise primer matching, even minor mismatches can cause failure |
| Workflow Sequence |
Library-then-capture or capture-then-library |
Common misconception about "capture-then-library" |
| Applications |
Useful for phylogenetic studies, methylation capture |
Suitable for point mutations, detects known mutations |
| Limitations |
Time-consuming |
Limited in detecting CNVs and SVs |
| Time Efficiency |
Typically longer, but can be reduced using rapid hybridization |
Faster library preparation |
| Starting DNA Quantity |
Requires more due to fragmentation losses |
Requires minimal template DNA |
| DNA Integrity |
Tolerates shorter fragment lengths, suitable for ancient DNA |
Challenging with short fragments |
| Methylation Capture |
Effective for cfDNA, suitable for early cancer screening |
Limited to samples with abundant gDNA |
| Suitability |
Flexible, sensitive, wide range of applications |
Rapid, efficient for targeted sequencing |
Conclusion
In summary, both capture-based and amplicon-based methods offer valuable capabilities for NGS, with each having specific strengths depending on the research context and objectives. The choice of method should be carefully considered, taking into account factors such as mismatch tolerance, reference genome availability, DNA quantity and integrity, and the type of genomic variations being studied. While capture-based methods may be more time-consuming, they offer significant advantages in terms of flexibility, sensitivity, and applicability to a wide range of genomic regions and sample types. Conversely, amplicon-based methods are more rapid and efficient for detecting known mutations, making them highly suitable for targeted sequencing applications.