

We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Microbial Genome-Wide Association Studies (mGWAS) is a new area of research that identifies genetic variation in microbial genomes associated with host variation or microbial phenotypes. CD Genomics is dedicated to genomic and population genetic analysis, and we use our experience in GWAS to help our clients understand how variation in microbial genomes affects host or pathogen phenotypes, such as drug resistance, virulence, host specificity, and prognosis.

We are dedicated to providing outstanding customer service and being reachable at all times.
The microbiome has emerged as an important non-genetic component influencing host phenotype and its impact on the host may go beyond the analysis of host genetics. Microbial Genome-Wide Association Studies (mGWAS) allows revealing the characteristics and mutations of microbiota associated with the host. mGWAS utilizes and develops human GWAS research methods to resolve the interactions and associations between variation (such as single nucleotide polymorphisms (SNP) and INDEL, gene presence-absence, copy number variation (CNV) and sequence inversions (SI)) and phenotypes in the microbial genome with the host and environment.
Our mGWAS service offers many great benefits and conveniences, such as a high-throughput bioinformatics pipeline and established reference database, as well as providing sequencing control at a cost per sample. The use of this strategy demonstrates a significantly different characterization of the microbiota and greatly deepens our understanding of its role in various body regions of human health and disease.
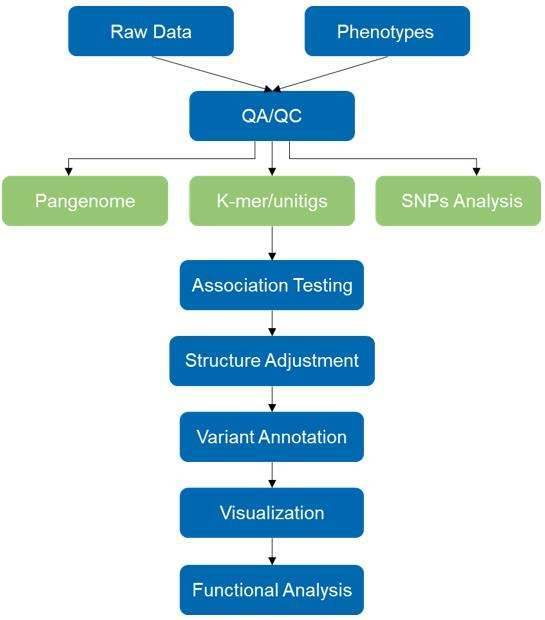
Our high-throughput mGWAS service can be used to advance
| ANALYSIS CONTENTS | DETAILS |
|---|---|
| Variant calling, evaluation and annotation | SNPs are called using HaplotypeCaller and filtered using VariantFiltration with parameter. |
| Enrichment of Gene Ontology (GO) term and KEGG pathway | Enrichment of Gene Ontology (GO) term and KEGG pathway are performed for candidate genes according to EnrichmentPipeline. |
| Definition of abundant and rare microbial OTUs | Briefly, the OTUs with relative abundances ≥0.1% across more than 50% of samples were defined as abundant OTUs (AT), whereas the OTUs with relative abundances <0.01% across more than 30% of samples but never abundant (≥0.1%) more than 30% samples were defined as rare OTUs (RT). |
| The microbial co-occurrence network analysis | Evaluation of correlation between host phylogenetic distance and target microbiota distance |

Specimens from human, animal, natural and industrial environments, as well as DNA and PCR products.
Deliverables: Raw sequencing data (FASTQ), trimmed and stitched sequences, quality-control dashboard, statistic data, and your designated bioinformatics report.
Reference
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to info@cd-genomics.com for inquiries.
Please fill out the form below: ×


Follow us on: