
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
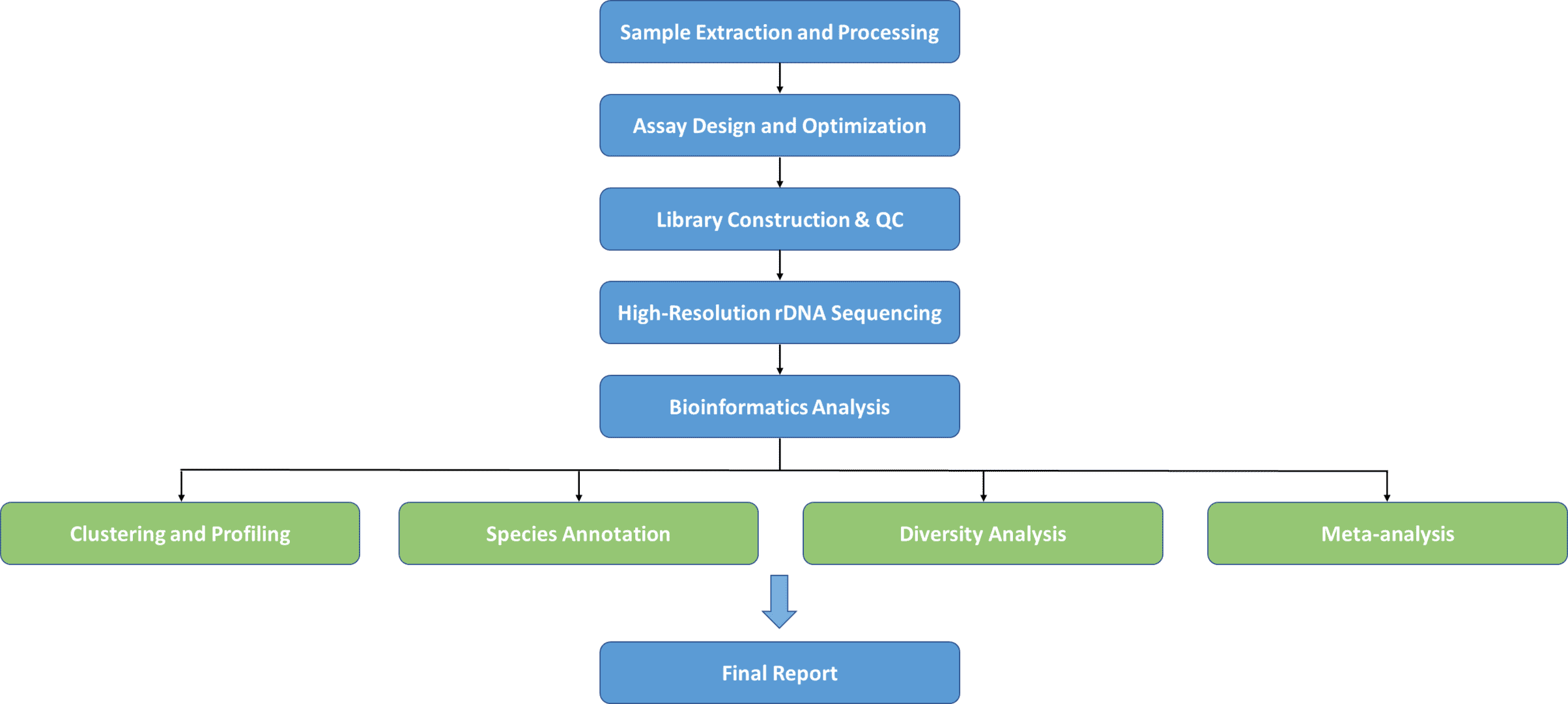
The specification for analyzing the structure of microbial diversity has been sequencing the rRNA gene. While it enables microbes to be identified at the species level, this process mainly does not offer adequate information to address communities at the sub-species level. For research of transmission or reliability or for exploring subspecies variance in disease connection, the species-level resolution is not sufficient. There are significant restrictions to strain level assessment using whole metagenome shotgun sequencing that can make it unfit for large-scale studies. Attaining adequate sequencing depth can be cost-prohibitive, and it is computationally very complicated to deconvolute complex communities like the oral microbiota even with proper coverage. High-resolution, yet cost-effective, high-throughput techniques for classifying microbial communities are therefore needed.

We are dedicated to providing outstanding customer service and being reachable at all times.

Between distinct species of microorganisms, containing nine hypervariable areas varying from about 30 to 100 base pairs long, the rRNA gene is highly preserved, varying significantly between bacteria. Highly conserved areas enable scientists to construct primer pairs that will augment the hypervariable area of their preference accurately and reliably in order to identify or characterize various bacterial communities. rRNA gene sequencing is the best instrument for studying bacterial and fungal taxonomy and molecular phylogeny, depending on the advancement of sequencing innovation. CD Genomics offers a full-length rRNA sequencing service to help assist your studies by taking advantage of PacBio SMRT long-reads sequencing methods.
The method provides numerous huge benefits, such as high throughput bioinformatic pipelines and reference databases founded, and low cost per specimen. The use of this strategy has indicated the microbiota's remarkably varied character and substantially developed our knowledge of its role in various body areas in human health and disease.
| Analysis Contents | Details |
|---|---|
| OTUs Clustering and Profiling | In genomics and metagenomics, it is an essential instrument that conducts taxonomic analysis of the microbial world by categorizing 16S rDNA amplicons into clusters called Operational Taxonomic Units (OTUs). With the support of Next Generation Sequencing (NGS) tools and grouping, it has become convenient for researchers to locate microbial diversity without cultivating the microbes in different situations. |
| Species Annotation | Classifying functional components along a genome's sequence. It enables DNA sequencing, which then generates sequences of unknown functions. |
| Meta-analysis | By sequencing association information from multiple research involving individual genetic loci or variants, meta-analysis enhances statistical significance. CD Genomics systematically analysed population-based case-control genetic association experiments in this meta-analysis to recognise all gene-specific SNVs related to the risk of osteosarcoma. |
| Alpha and Beta Diversity | We quantify alpha-diversity within a particular habitat type as the observed richness (number of taxa) or evenness (relative abundances of those taxa) of an average sample. We evaluate beta-diversity among specimens within a habitat as the variance in the concentration of the group (the identity of taxa discovered). |

Deliverables: Raw sequencing data (FASTQ), trimmed and stitched sequences, quality-control dashboard, statistic data, and your designated bioinformatics report.
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to info@cd-genomics.com for inquiries.
Please fill out the form below: ×


Follow us on: