
 Sample Submission Guidelines
Sample Submission Guidelines
What is Immune Repertoire
The adaptive immune system plays an extremely important role in counteracting infectious pathogens and shielding the human body. The foundation of the adaptive immune system is fundamentally reliant upon the enormous diversity of T cell receptors and other immune receptors that can recognize epitopes from an astronomical number of different exogenous antigens and endogenous host factors.
Immune repertoire is known as the collection of functionally diverse immune cells in the circulatory system, which is a key measure of immunological complexity. T cells and other immune cells respectively orchestrate cellular and humoral immune responses in the organism. They each function via their surface receptors to recognize and bind to antigens, playing a critical role in eliminating pathogens or intrabody tumor cells. Operating independently, T cells and these other immune cells mediate the body's complementary cellular and humoral immune responses, each utilizing their specific receptors on their surface. These receptors facilitate the identification and binding of antigens, thereby playing a fundamental role in the clearance of pathogens or endogenous tumor cells. The study of immune repertoire is crucial to understanding the response of adaptive immunity, and ultimately, sheds light on the discovery of novel infectious agents and holds the invaluable potential to aid in antibody or vaccine development, clinical diagnosis, treatment, and prevention.
If you want to learn more about the introduction of Immune Repertoire Sequencing, please check out our article "Immune Repertoire Sequencing: Introduction, Workflow, and Applications".
High Throughput Immune Repertoire Sequencing
The tradition strategies for studying the immune repertoire, which includes spectratyping, Sanger sequencing et al., are laborious, costly, and are insufficient for generating a high-resolution picture of the immune repertoire. NGS (next generation sequencing) offers a powerful approach capable of analyzing the immune repertoire in a high-throughput manner. By providing enormous sequencing data, NGS has created an unprecedentedly high-resolution picture of the immune repertoire, which provides great potential for revealing dynamic changes in clonal populations during antigen stimulation.
Immune Repertoire Sequencing - Target Regions
In the realm of immune repertoire sequencing, the focus lies on T/B lymphocytes. Employing techniques such as multiplex PCR or 5'RACE, the objective is to amplify the complementary determining regions (CDR3) that determine the diversity of B cell receptors or T cell receptors. CDR3, being the most variable region, stands as the epicenter of exploration in immune repertoire sequencing studies. Its prominence is underscored by its status as the most extensively researched domain within the field.
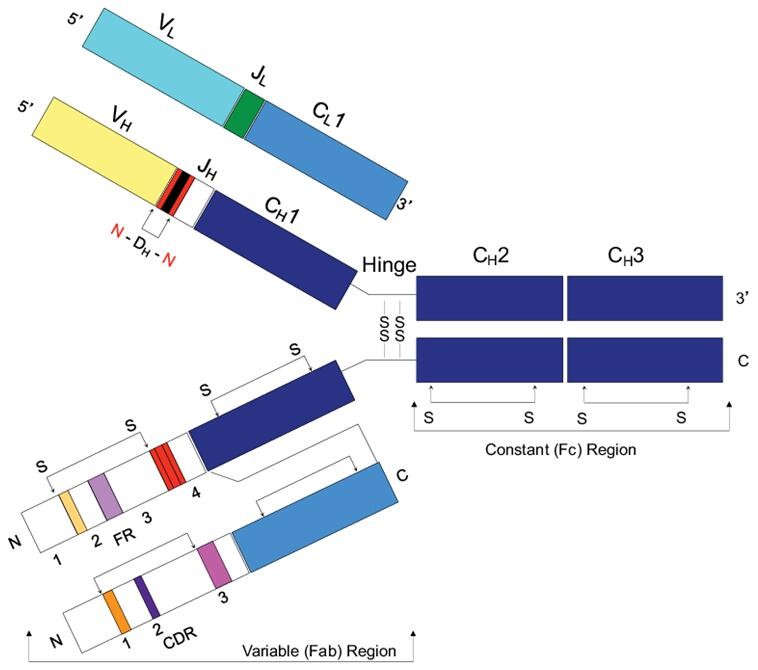 Figure 1. IgG structure (Brian Douglas Weitzner 2015)
Figure 1. IgG structure (Brian Douglas Weitzner 2015)
What is BCR Sequencing
BCR sequencing employs high-throughput sequencing to detect the targeted amplification of BCR heavy and light chains. It comprehensively analyzes the base sequences resulting from BCR gene rearrangements, along with the abundance of each sequence. This technique finds application in investigating the transcriptional profiles and interrelationships of different B cell clones, delving into the functional specificity of B cells. Consequently, it aids in elucidating phenomena such as humoral immune response tolerance and the role of high-frequency mutations in abnormal antigen recognition by B cells.
The applications of BCR-Seq encompass:
- Screening for somatic mutations in cellular populations of patients with primary immunodeficiency diseases like isolated IgA deficiency.
- Guiding the treatment of autoimmune diseases (e.g., SLE, multiple sclerosis, type 1 diabetes), and monitoring disease progression during treatment.
- Assessing the efficacy and tolerance of drug treatments for infectious diseases and malignant tumors.
- Predicting and intervening in transplant rejection reactions, providing guidance on tolerance and rejection responses.
What is TCR Sequencing
T-cell receptor sequencing (TCR-Seq) utilizes high-throughput sequencing (HTS) to detect the targeted amplification of T-cell antigen recognition determinant surface molecules. It analyzes their diversity, along with the TCR gene rearrangement base sequences and the abundance of each sequence before and after T-cell antigen recognition. This technique reflects changes in the cellular immune response mediated by T cells in physiological and pathological states. It is instrumental in studying the transcriptional profiles and interrelationships of different T cell clones, thereby unveiling deeper levels of T cell functional specificity.
Applications of TCR-Seq include:
- Detection and guidance of post-tumor immunotherapy monitoring and treatment.
- Evaluation of the effectiveness and tolerance of anti-infective immunotherapy.
- Prediction and guidance of acute rejection in transplant rejection reactions.
- Clinical trials and scientific research on autoimmune diseases.
- Biomarkers and diagnostics for various infectious and neuroplastic diseases.
TCR Sequencing Service
CD Genomics has developed advanced TCR Sequencing strategy to amplify and sequence the immune repertoire with Next Generation Sequencing (NGS). Now, we are providing a comprehensive solution to evaluate the diversity of immune repertoire precisely in a cost-effective way by pooling the samples. We can tailor library construction and sequencing protocols to meet the specific requirements of different sample types and research projects, ensuring a personalized approach for our clients. Additionally, we integrate cutting-edge bioinformatics algorithms to facilitate visual data analysis, aligning with world-leading standards in computational biology.
Our Immune Repertoire Sequencing strategy is to capture the complementary determining region (CDR) of T-cell receptor (TCR) using multiple-PCR or 5' RACE methods, followed by deep sequencing. The workflow for the strategy is illustrated in the Figure below.
Successful construction of TCR libraries can be achieved using samples obtained by immunomagnetic bead sorting of CD3-positive T cells and Ficoll-Paque gradient centrifugation separation of peripheral blood lymphocytes.
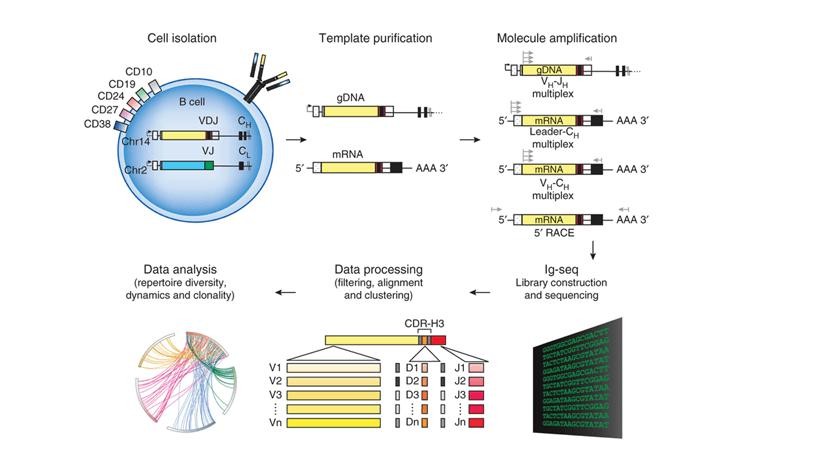 Figure 2. Schematics of steps for high-throughput sequencing of the Ig sequence repertoire (Georgiou et al., 2014).
Figure 2. Schematics of steps for high-throughput sequencing of the Ig sequence repertoire (Georgiou et al., 2014).
Selection of Two PCR Techniques
| PCR Technique | Multiplex PCR | 5'RACE |
| Principle | Same as regular PCR | Rapid amplification of cDNA's 5' end in low-abundance transcripts |
| Amplification Region | CDR3 | CDR1-3 |
| Sample Type | DNA, RNA | RNA (C region known required) |
| Advantages | Simple operation, complete data | Minimizes PCR amplification bias to a great extent |
| Disadvantages | PCR amplification bias | Laborious operation, loss of partial sequences, lower repeatability compared to multiplex PCR |
Note: Choose different sequencing platforms and modes based on different needs.
What are the Advantages of Our Immune Repertoire Sequencing
- Comprehensive One-stop Service: We provide professional services covering the entire process, from experimental design, sample nucleic acid extraction and amplification, library construction, sequencing, information analysis, to data mining.
- Cutting-edge Amplification Techniques: Utilizing multiplex PCR or 5'RACE technology, we employ primers optimized through plasmid adjustments, significantly reducing biases and achieving equivalent amplification for various clones.
- High reproducibility: High consistency of clones when the same sample is prepared into a library twice.
- More accurate clonal quantification: Aligning low-quality reads with high-quality clones to recover important data, ensuring no loss of clonal quantification information.
- Enhanced error correction and mismatch handling: Using multi-layer clustering methods to correct errors introduced by PCR and sequencing.
What are the Application of Immune Repertoire Sequencing
- Tumor Therapy: There is research into infiltrating lymphocytes present in various types of tumors (including colorectal, ovarian, liver cancer, and melanoma), tracking shifts in the immunological microenvironment at the onset of tumor formation. This aids in pinpointing targets for immunotherapy and thereby bolstering the anti-tumor response and effectiveness of such treatments.
- Transplantation and Immune Reconstitution: During organ or bone marrow transplantation, there is often the induction of a host rejection response which can lead to chronic graft versus host disease. At these times, immunoglobulin sequencing technology can be employed to probe the post-transplant T-cell repertoire, facilitating identification of high-risk groups for postoperative infection and relapses.
- Autoimmune Diseases: Researchers analyze aberrantly expressed immune cells (T cells or antibodies) and as such, discover diagnostic biomarkers. The quest for biomarkers surpassing current standards and investigating immunological mechanisms pertaining to autoimmunity can thus provide substantive foundation for disease diagnosis and treatment.
- Medication and Vaccine Evaluation: Assessment of peripheral blood samples post-medication for disease, verifies whether the drug triggers an immune response and its efficacy.
- Other Aspects: Immunoglobulin libraries play a critical role in the study of infectious diseases, mapping differential immune spectra, and investigations related to immunotherapeutic modalities.
Immune Repertoire Sequencing Workflow
Service Specification
Sample Requirements
|
|
 |
Sequencing Strategy
|
 |
Bioinformatics Analysis
|
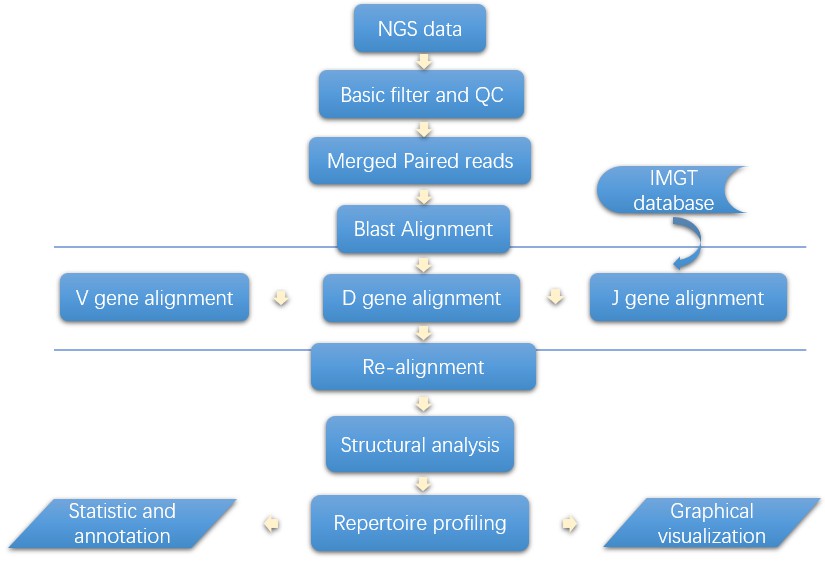
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Immune Repertoire Sequencing for your writing (customization)
Reference:
- Gravina, S et al. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biology, 2016 17(1):150.
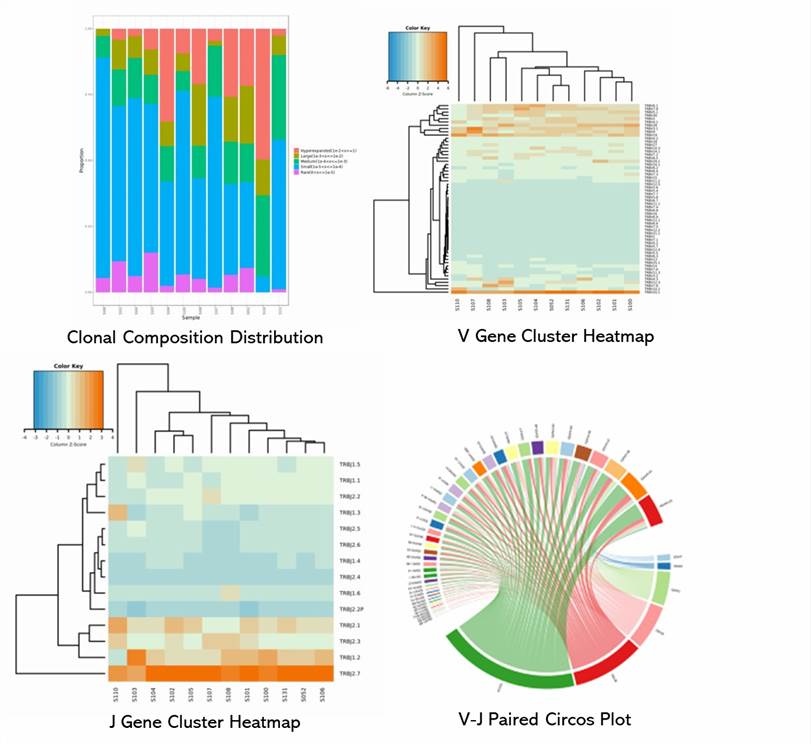
1. What distinguishes the encoding genes of TCR and BCR chains? Is there a recommended chain?
The Beta chains of T-Cell Receptors (TCR) and the Heavy chains of B-Cell Receptors (BCR) are encoded by variable (V), diversity (D), and joining (J) genes, whereas the Alpha chains of TCR and Light chains of BCR are encoded solely by V and J genes. A review of the current scientific literature suggests a higher prominence of research focused on the Beta chain of TCR and the Heavy chain of BCR. Consequently, these may be recommended for relevant studies, although the choice should ultimately be project and hypothesis-driven.
2. Can immunoglobulin repertoire sequencing distinguish between IgG, IgM, IgD, and IgE?
By sequencing the constant (C) region, the isoforms of immunoglobulins (IgG, IgM, IgD, and IgE) can be clearly delineated. The requisite primers for such differentiation are available. However, it is imperative that the samples should be RNA. It is accomplished through a method of multiplex PCR that utilizes primers from the variable (V) region to the constant (C) region. The distinguishing sequence is approximately 30 base pairs, resulting in products that are around 200-300 base pairs in length.
3. For immune repertoire sequencing, is it better to use genomic DNA or RNA as a template?
In the context of immune repertoire sequencing, both genomic DNA (gDNA) and RNA serve distinct roles and bring their own set of advantages and disadvantages:
When using gDNA as the template material, the benefits include an accurate reflection on the clone count of immune cell receptors, due to the fact that each gene has only two copies. Not to mention, DNA is a stable molecule that eases the process of storage. However, a potential shortcoming of using gDNA lies in its low copy number, which may require an elevated amount of sample material. Additionally, the large intron region between the J and C regions might hinder the amplification of the full-length CDR due to sequencing length limitations.
On the other hand, using RNA allows the design of primers in the C region which enables the amplification of the full-length CDR, thanks to there being no intron between the J and C regions. Owing to its high levels of expression, the template quantity from RNA is generally high, therefore less sample material might be needed. However, it should be noted that the clonality of immune cell receptors can be influenced by mRNA expression levels, possibly precluding an objective reflection of the actual clone count. Also, RNA is less stable than DNA, which requires stringent procedures for sample preservation and handling.
4. What information can be obtained from IR-seq?
The application of immunoglobulin or receptor sequencing (IR-seq) can furnish considerable insights into the heterogeneity and clonality of T and B cell populations within an individual. IR-seq enables the identification of specific T-cell receptor (TCR) or B-cell receptor (BCR) sequences, facilitating the assessment of clonal expansion, allowing for a comprehensive understanding of the repertoire, uncovering changes in response to stimuli, be it infection, vaccination, or manifestations of autoimmune diseases.
5. What are the main steps involved in IR-seq?
Executing IR-seq is a multi-phase process. It begins with the collection of the sample, generally either peripheral blood or a tissue biopsy. The extraction of RNA or DNA ensues, followed by the amplification of TCR or BCR genes, achieved through methods like PCR or targeted enrichment. This is promptly succeeded by high-throughput sequencing. The final step entails bioinformatic analysis, with an aim to identify and quantify unique TCR or BCR sequences present.
6. What are the key applications of IR-seq?
Immune Repertoire Sequencing (IR-seq) is a seminal tool, with wide-ranging applications in both immunology and clinical research. It is assiduously deployed in the exploration of immune responses against external factors such as pathogens and tumors, as well as internal challenges like autoimmune disorders. Moreover, its efficacy extends to monitor events like transplants to determine instances of organ rejection, and evaluate the diversity and senescence of the immune system.
7. What are some emerging technologies in IR-seq?
In the constantly evolving field of IR-Sequencing, several progressive technologies are establishing their footing. Notably, single-cell IR-seq is one of them, which proffers the capability to profile individual immune cells. Another is the spatially-resolved IR-seq that facilitates the mapping of immune repertoires within specific tissue regions. Furthermore, the advent of machine learning and artificial intelligence is significantly bolstering the analysis of voluminous IR-seq datasets, aiding in distilling meaningful, potentially breakthrough insights.
Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire
Journal: Nature genetics
Impact factor: 27.125
Published: 03 April 2017
Background
The adaptive immune system's effectiveness against infections depends on T cells producing diverse αβ-heterodimeric T cell receptors (TCRs) via V(D)J recombination. TCR specificity arises from sequence diversity, particularly in the CDR3 region. T cells proliferate upon antigen recognition, forming memory populations with identical TCR rearrangements. Despite vast TCRβ diversity, public T cell responses occur when certain TCRβ chains are shared among individuals, especially in response to common antigens. Public T cell responses are observed in various infectious diseases and conditions like malignancies and autoimmunity. High-throughput sequencing of TCRβ chains allows for the identification of significant associations between TCR sequences and specific phenotypes, offering potential diagnostic applications for immune-related conditions.
Methods
- Human peripheral blood samples
- Fred Hutchinson Cancer Research Center Research Cell Bank
- Genomic DNA extraction
- Immunosequencing
- Illumina HiSeq system
- Identification of phenotype-associated TCRs
- Inference of phenotype
- Assessing classification performance
Results
The study aimed to identify CMV-associated TCRβ sequences by analyzing peripheral blood samples from 641 subjects with known CMV serostatus. Through statistical analysis, 164 TCRβ chains were found to be significantly enriched among CMV+ subjects. Subsequent investigation of HLA restriction revealed that 45 of these TCRβ chains were associated with specific HLA-A or HLA-B alleles. Comparison with previously reported CMV-reactive TCRβ sequences showed that while some overlapped, many did not display differential incidence between CMV+ and CMV- subjects in this cohort. Further analysis incorporating both presence/absence and abundance metrics identified additional CMV-reactive TCRβ sequences, suggesting the effectiveness of this approach in identifying relevant TCRβ sequences associated with CMV infection.
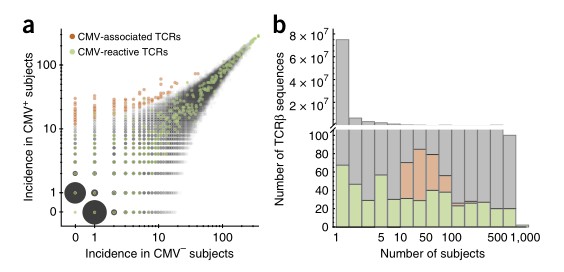 Fig 1. Identification of CMV-associated TCRβs.
Fig 1. Identification of CMV-associated TCRβs.
This study controlled for repertoire sampling depth and observed a distinct association between the number of CMV-related TCRβ sequences and CMV serostatus. The investigators constructed a binary classifier to predict CMV serostatus based on the presence or absence of CMV-specific TCRβ sequences, achieving remarkable accuracy (an AUROC of 0.99 for all data and 0.93 for cross-validation). Further validation performed on an independently collected cohort confirmed the robustness of the classifier, evidenced by an AUROC of 0.94 and demonstrating high sensitivity and specificity. These findings thus illustrate the noteworthy diagnostic potential of this approach.
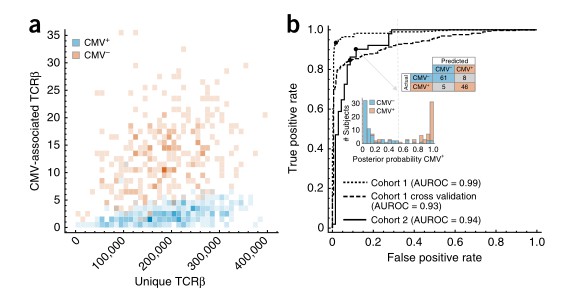 Fig 2. The incidence of CMV-associated TCRβs is diagnostic of CMV serostatus.
Fig 2. The incidence of CMV-associated TCRβs is diagnostic of CMV serostatus.
Conclusion
The study investigated whether pathogen exposure signatures could be detected by analyzing public T cell receptors (TCRs) in the T cell repertoire of 666 subjects with known CMV serostatus. They developed a statistical framework to diagnose CMV status accurately, achieving high specificity and sensitivity in both the original and validation cohorts. Additionally, they identified CMV-associated TCRβ molecules and accurately predicted HLA-A and HLA-B alleles. The approach has broad potential for diagnosing various disease states and immunological phenotypes, offering a highly parallelizable diagnostic strategy based on the common format of somatic TCR recombination.
Reference:
- Emerson R O, DeWitt W S, Vignali M, et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nature genetics, 2017, 49(5): 659-665.
Here are some publications that have been successfully published using our services or other related services:
The HLA class I immunopeptidomes of AAV capsid proteins
Journal: Frontiers in Immunology
Year: 2023
Isolation and characterization of new human carrier peptides from two important vaccine immunogens
Journal: Vaccine
Year: 2020
Change in Weight, BMI, and Body Composition in a Population-Based Intervention Versus Genetic-Based Intervention: The NOW Trial
Journal: Obesity
Year: 2020
Sarecycline inhibits protein translation in Cutibacterium acnes 70S ribosome using a two-site mechanism
Journal: Nucleic Acids Research
Year: 2023
Identification of a Gut Commensal That Compromises the Blood Pressure-Lowering Effect of Ester Angiotensin-Converting Enzyme Inhibitors
Journal: Hypertension
Year: 2022
A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells
Journal: Cells
Year: 2021
See more articles published by our clients.


