




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
DNA methylation, a crucial epigenetic modification, modulates gene expression without altering the DNA sequence. It plays a fundamental role in numerous biological processes such as organism growth and development, cell differentiation, and disease occurrence and progression. Genome-wide bisulfite sequencing (WGBS) technology has emerged as a powerful tool in this context. By treating unmethylated cytosine with bisulfite to convert it into uracil while leaving methylated cytosine unchanged, and integrating with high-throughput sequencing, WGBS enables the construction of methylation maps with single-base resolution across the entire genome. This technology has opened new avenues for in-depth exploration of DNA methylation functions and mechanisms, demonstrating extensive application potential across various life science disciplines.
Service you may intersted in
If you want to learn more about the WGBS, please refer to:
Embryonic development: In the process of embryonic development, DNA methylation plays a vital role, which is involved in key processes such as cell fate determination and lineage differentiation. WGBS technology can map the whole genome methylation at different stages of embryonic development and reveal the dynamic change law of methylation. For example, in the study of mouse embryo development, WGBS found that large-scale demethylation occurred in the genome after fertilization, and then there was a process of demethylation before and after implantation. These methylation changes are closely related to the totipotency, pluripotency and differentiation of embryonic cells. By analyzing the methylation status of specific gene promoter region, we can deeply understand the mechanism of gene expression regulation during embryonic development, and provide theoretical basis for studying diseases caused by abnormal embryonic development.
Cell differentiation: Cell differentiation is the basis of individual development of multicellular organisms, and DNA methylation plays a key role in it. WGBS can be used to study the methylation differences of different types of cells during differentiation. Taking the differentiation of hematopoietic stem cells as an example, through WGBS analysis of hematopoietic stem cells and various blood cells formed by differentiation, it was found that the methylation pattern of genome changed significantly with the differentiation. The promoter region of some genes related to stem cell self-renewal gradually methylates during the differentiation process, which leads to gene expression silence, while the methylation level of genes related to specific functions of blood cells decreases correspondingly, which promotes gene expression. This indicates that WGBS can reveal the regulatory network of gene expression mediated by methylation in the process of cell differentiation, which is helpful to understand the molecular mechanism of cell differentiation.
Research on Tumor
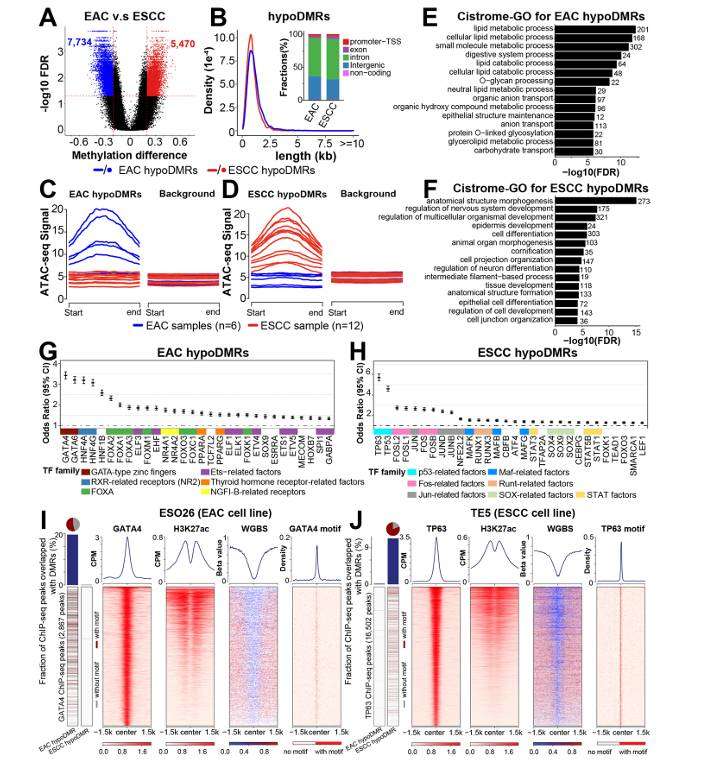 Subtype-specific differentially methylated regions in esophageal cancer (Zheng et al., 2023)
Subtype-specific differentially methylated regions in esophageal cancer (Zheng et al., 2023)
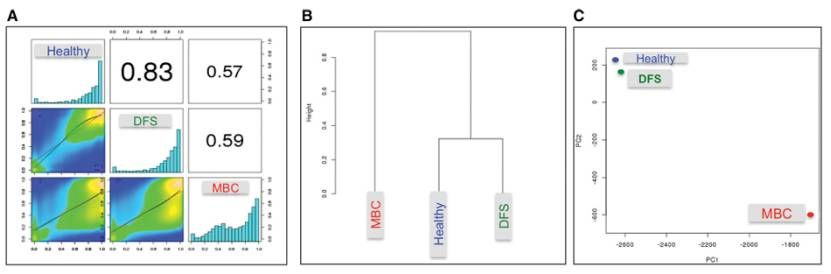 WGBS reveals that MBC methylation profiles differ from DFS and H (Christophe et al., 2015)
WGBS reveals that MBC methylation profiles differ from DFS and H (Christophe et al., 2015)
Neurodevelopmental diseases
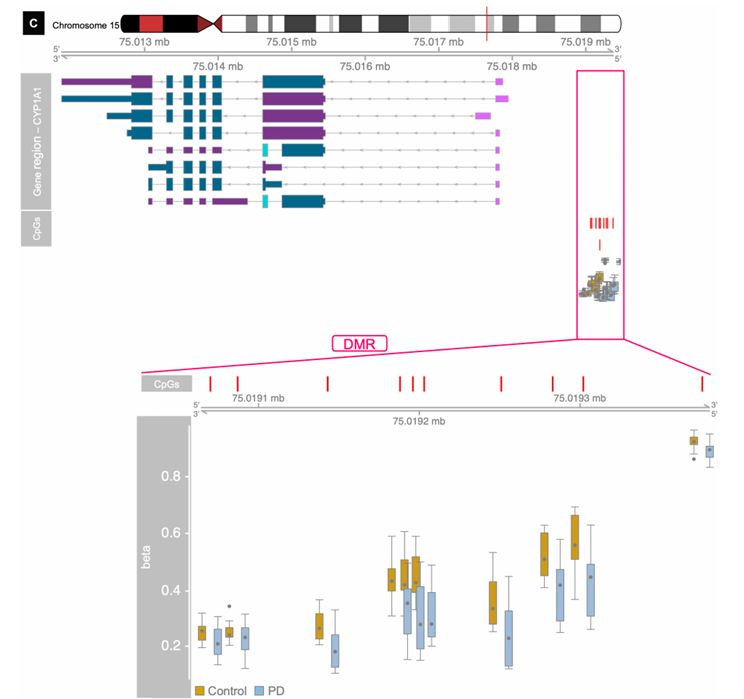 Differentially methylated regions nearest to the CYP1A1 gene (Adrienne R et al., 2021)
Differentially methylated regions nearest to the CYP1A1 gene (Adrienne R et al., 2021)
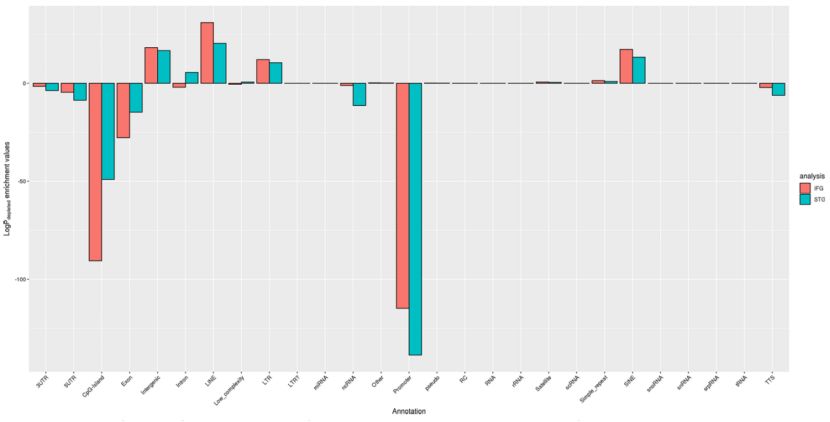 Enrichment of genomic features among the differentially methylated regions (Li et al., 2020)
Enrichment of genomic features among the differentially methylated regions (Li et al., 2020)
Cardiovascular disease
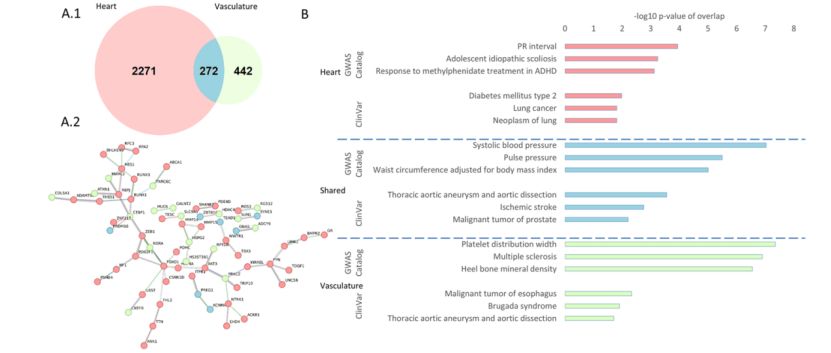 Methylation sites in heart and vasculature-related studies (Mykhailo et al., 2023)
Methylation sites in heart and vasculature-related studies (Mykhailo et al., 2023)
Crop genetic improvement: In crop breeding, it is of great significance to understand the methylation pattern of crop genome for genetic improvement. WGBS can be used to analyze the methylation differences of different crop varieties or the same variety under different environmental conditions. For example, in rice research, through WGBS of different rice varieties, it was found that the methylation status of some genes related to important agronomic traits such as yield, quality and stress resistance was different. These methylation differences may affect gene expression, and then lead to trait differences. Through the study of these methylation sites, we can develop molecular markers closely linked with excellent traits for molecular marker-assisted breeding and improve breeding efficiency. In addition, studying the methylation changes of crops in response to adversity (such as drought, salinity, etc.) is helpful to explore the genes related to stress resistance and their regulatory mechanisms, and provide theoretical support for cultivating crop varieties with strong stress resistance.
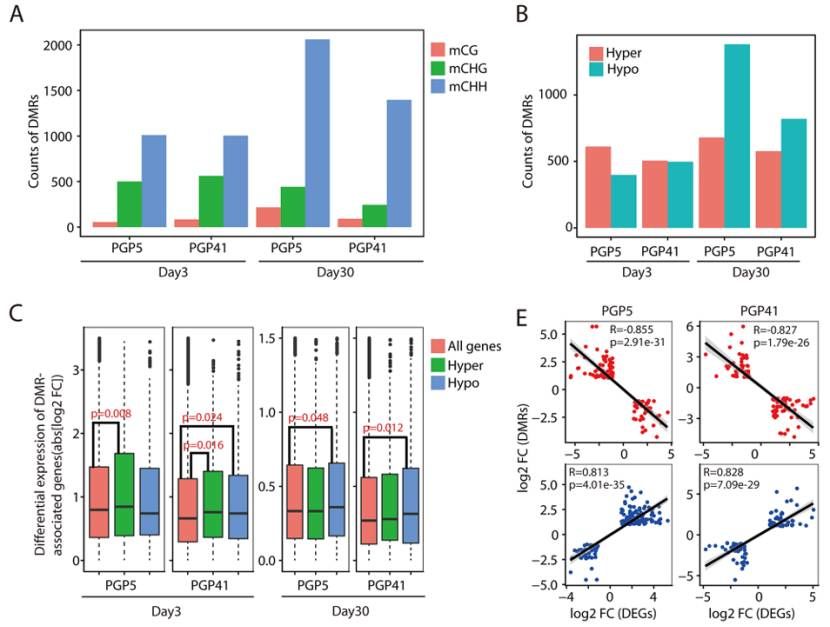 Variation in DNA methylome profiles and correlations with gene expression (Chen et al., 2022)
Variation in DNA methylome profiles and correlations with gene expression (Chen et al., 2022)
Livestock and poultry genetic breeding: In livestock and poultry breeding, WGBS technology can also be used in genetic breeding research. Through WGBS analysis of genomes of different breeds of livestock and poultry or different individuals of the same breed, methylation sites related to important economic traits such as growth performance, meat quality and reproductive performance can be found. For example, in the breeding research of pigs, it is found that the methylation level of some genes related to muscle growth and fat metabolism is closely related to the growth speed and meat quality of pigs. Using these methylation information, we can establish a new method for genetic evaluation of livestock and poultry, and realize accurate selection and genetic improvement of excellent traits of livestock and poultry.
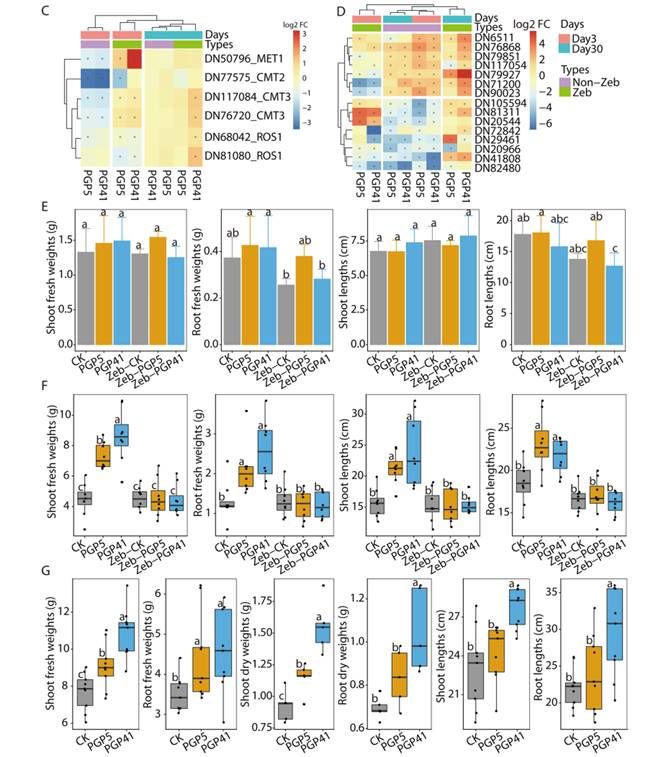 Schematic representation of the two-step interaction between PGPB and plants mediated by DNA methylation and root recruitment (Chen et al., 2022)
Schematic representation of the two-step interaction between PGPB and plants mediated by DNA methylation and root recruitment (Chen et al., 2022)
Microbial infection and host interaction: In the process of pathogen infecting host, the DNA methylation of microorganism itself will change dynamically. Taking Helicobacter pylori infection in human stomach as an example, the researchers found that the methylation status of some virulence-related genes of Helicobacter pylori changed during the infection process by using WGBS technology. The methylation level of promoter regions of some genes decreased, which increased the expression of these genes and enhanced the colonization ability and pathogenicity of Helicobacter pylori. At the same time, the DNA methylation pattern of host cells will also be affected. Through WGBS analysis of gastric mucosal cells infected by Helicobacter pylori, it was found that the methylation level of some genes related to immune response and cell proliferation changed, which further affected the immune response and cell physiological function of the host.
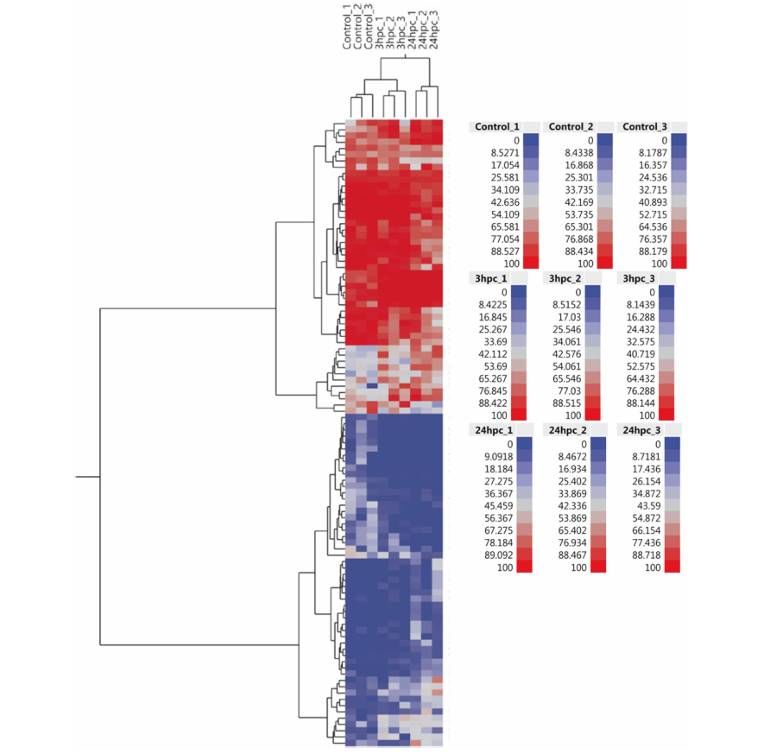 Heat map and hierarchical cluster analysis of the top 100 differentially methylated CpG identified in the study (Basavaraj et al., 2019)
Heat map and hierarchical cluster analysis of the top 100 differentially methylated CpG identified in the study (Basavaraj et al., 2019)
Symbiotic microorganism-host epigenetic relationship: There are complex interactions between symbiotic microorganisms and their hosts. For example, beneficial bacteria such as Bifidobacterium in the intestine form a symbiotic relationship with the host. Using WGBS technology to study the intestinal epithelial cells and symbiotic microorganisms of the host, it is found that symbiotic microorganisms can affect the physiological processes such as metabolism and immunity of the host by regulating the DNA methylation pattern of the host cells. At the same time, the physiological state of the host will also feedback the DNA methylation of symbiotic microorganisms, thus regulating their growth, metabolism and interaction with the host.
Environmental microbial community methylation: In complex environments such as soil and ocean, the function and ecology of microbial communities are very important for maintaining ecological balance. WGBS technology can be used to analyze the DNA methylation characteristics of environmental microbial communities. For example, in the study of soil microbial community, through WGBS of microorganisms in different fertility soils, it is found that there are specific methylation patterns of microbial groups related to nutrient cycle and environmental adaptation. In high fertility soil, the methylation level of some key genes of microorganisms involved in nitrogen and phosphorus cycling is low, and the gene expression activity is high, which is beneficial to improve the availability of soil nutrients.
Industrial fermentation microbial community epigenetic regulation: In the process of industrial fermentation, the synergistic effect of microbial communities determines the fermentation efficiency and product quality. WGBS technology is helpful to reveal the epigenetic regulation mechanism among members of industrial fermentation microbial community. Taking alcohol production by Saccharomyces cerevisiae as an example, it was found that the DNA methylation pattern of yeast cells changed at different stages of fermentation. At the initial stage of fermentation, the methylation level of genes related to sugar metabolism is low, which promotes the uptake and metabolism of sugar by yeast. In the late stage of fermentation, the methylation status of some genes related to cell stress response changed to adapt to the changes of fermentation environment.
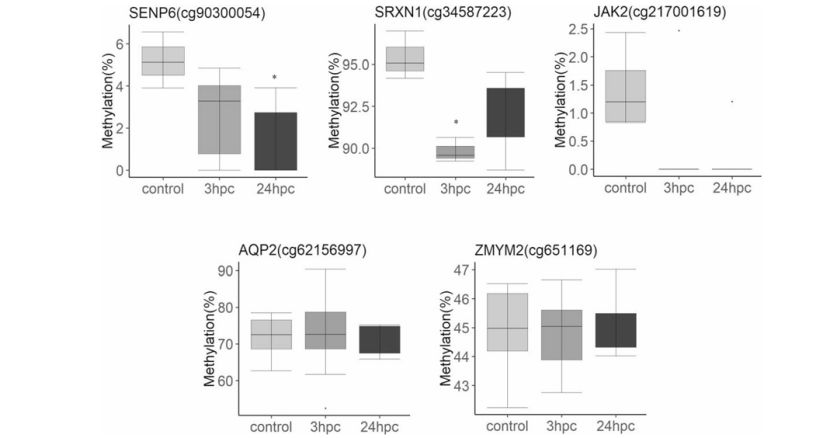 Pyrosequencing validation of CpG sites identified in the genome wide methylation including SENP6 (cg90300054), SDF4 (cg63545568), JAK2 (cg217001619), SRXN1 (cg34587223), ZMYM2 (c651169) (Basavaraj et al., 2019)
Pyrosequencing validation of CpG sites identified in the genome wide methylation including SENP6 (cg90300054), SDF4 (cg63545568), JAK2 (cg217001619), SRXN1 (cg34587223), ZMYM2 (c651169) (Basavaraj et al., 2019)
Despite the remarkable achievements of WGBS in various research fields, it still faces several challenges. Firstly, the high cost associated with the technology, from sample processing to sequencing analysis, requires professional equipment and a large amount of reagents, limiting its large-scale application. Secondly, the complexity of data analysis demands professional bioinformatics knowledge and high-performance computing resources to interpret the massive data generated. Additionally, strict sample requirements imply that the quality and quantity of samples can impact the accuracy of experimental results.
However, with the continuous development of technology, these challenges are expected to be gradually overcome. In the future, WGBS technology may make breakthroughs in the following aspects. On the one hand, technical optimization will reduce the cost and improve the efficiency and accuracy of the experiment. On the other hand, the integration of multi-omics will become a trend, and the combination of WGBS with transcriptomics, protein omics and other technologies can reveal the molecular mechanism of diseases more comprehensively. In addition, the development of single cell WGBS technology will contribute to the in-depth study of cell heterogeneity and provide more powerful support for accurate diagnosis and treatment of diseases.
In conclusion, WGBS technology has demonstrated substantial application potential in biological development, disease research, agricultural breeding, and microbiology due to its high resolution and genome-wide coverage. Despite the existing challenges of high cost and complex data analysis, continuous technological progress and innovation are expected to position WGBS as an indispensable tool in life science research in the future. It will provide crucial technical support for unraveling the mysteries of life, combating major diseases, and promoting agricultural development, thereby opening a new chapter in life science research.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.