




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Hi-C technology overcomes the limitations of conventional genomics, which is constrained to linear sequences, by converting the spatial interactions of chromatin information into sequencable DNA molecules. This technology provides a revolutionary tool for analyzing the structure-function relationship of three-dimensional genomes. In this paper, we systematically discuss the challenges and solutions of Hi-C experiments, from sample preparation to data generation, by focusing on the in-depth integration of technical principles and biological applications.
The article further integrates the intersection of molecular biology, bioinformatics, and clinical medicine to analyze the high complexity of Hi-C technology. The technology's intricacies are examined at several levels, from the control of chemical kinetics of in-situ cross-linking to the optimization of the specificity of enzyme ligation and the dynamic balance between the sequencing read length and the coverage. At each stage of the operation, a precise trade-off between the sensitivity of the technology and the background noise is required. To address this characteristic, this paper proposes a "prevention-detection-correction" quality control system and illustrates the scientific logic of the dynamic adjustment of experimental parameters based on the suitability of clinical samples and special biomaterials (e.g., plant cell wall penetration, FFPE tissue repair). By examining technical impediments and pioneering approaches, the paper establishes a methodological cornerstone for the translation of 3D genomic research from fundamental theory to clinical diagnosis and targeted therapy.
The three-dimensional spatial configuration of the genome has emerged as a pivotal research focus in the life sciences over the past decade. Conventional genomics studies have centered on the linear sequence information of DNA; however, the advent of Hi-C technology has precipitated a paradigm shift, empowering scientists to elucidate the spatial interaction network of chromosomes within the nucleus in a high-throughput manner. This technology has not only revealed chromosomes from nanoscale chromatin loops to macro-scale compartmentalized hierarchical folding patterns, but also provided a novel perspective for resolving gene regulation, disease mechanisms, and evolutionary processes. For instance, in cancer research, Hi-C technology has successfully revealed how aberrant rearrangements of the three-dimensional genome in tumor cells lead to aberrant activation of proto-oncogenes, providing a theoretical basis for targeted epigenetic therapy.
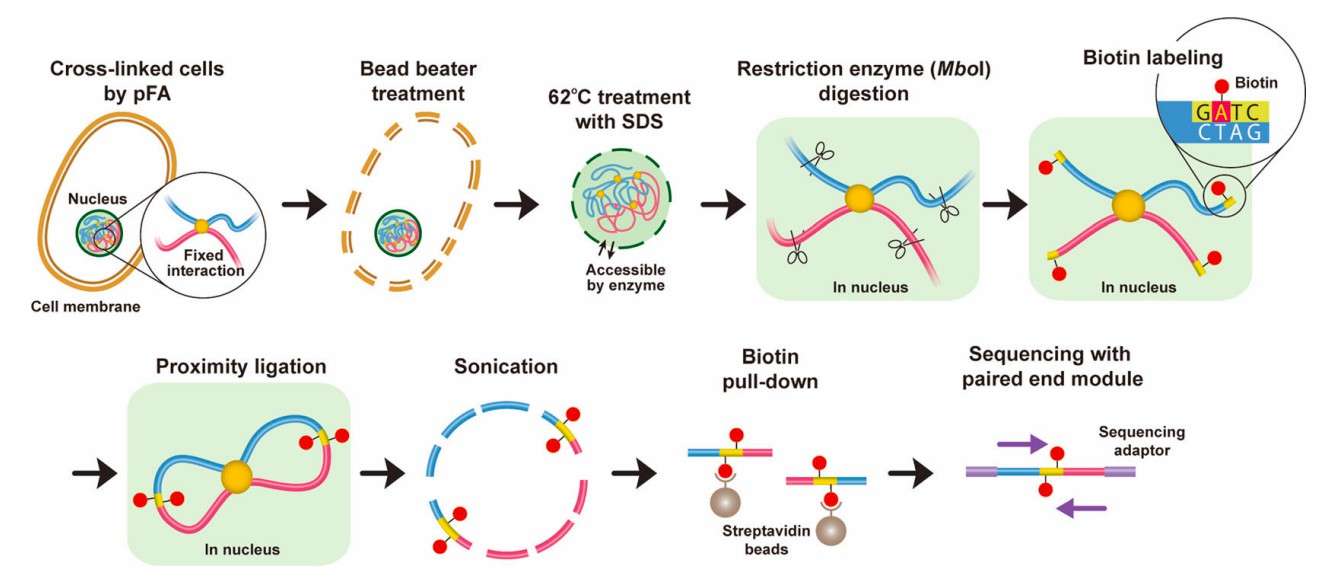 Hi-C experimental procedure (Tanizawa et al., 2025)
Hi-C experimental procedure (Tanizawa et al., 2025)
However, Hi-C experiments are more complex than conventional sequencing technologies, and the quality of their data is contingent on the rigor of the experimental process. Small deviations in each step of the operation, from the control of chemical kinetics of cell cross-linking to the molecular precision of chromatin zymography, may introduce systematic errors or even mask the real biological signals. Consequently, the establishment of standardized operation procedures and a comprehensive understanding of the principles underlying these procedures have become imperative for Hi-C technology to progress from fundamental research to clinical translation.
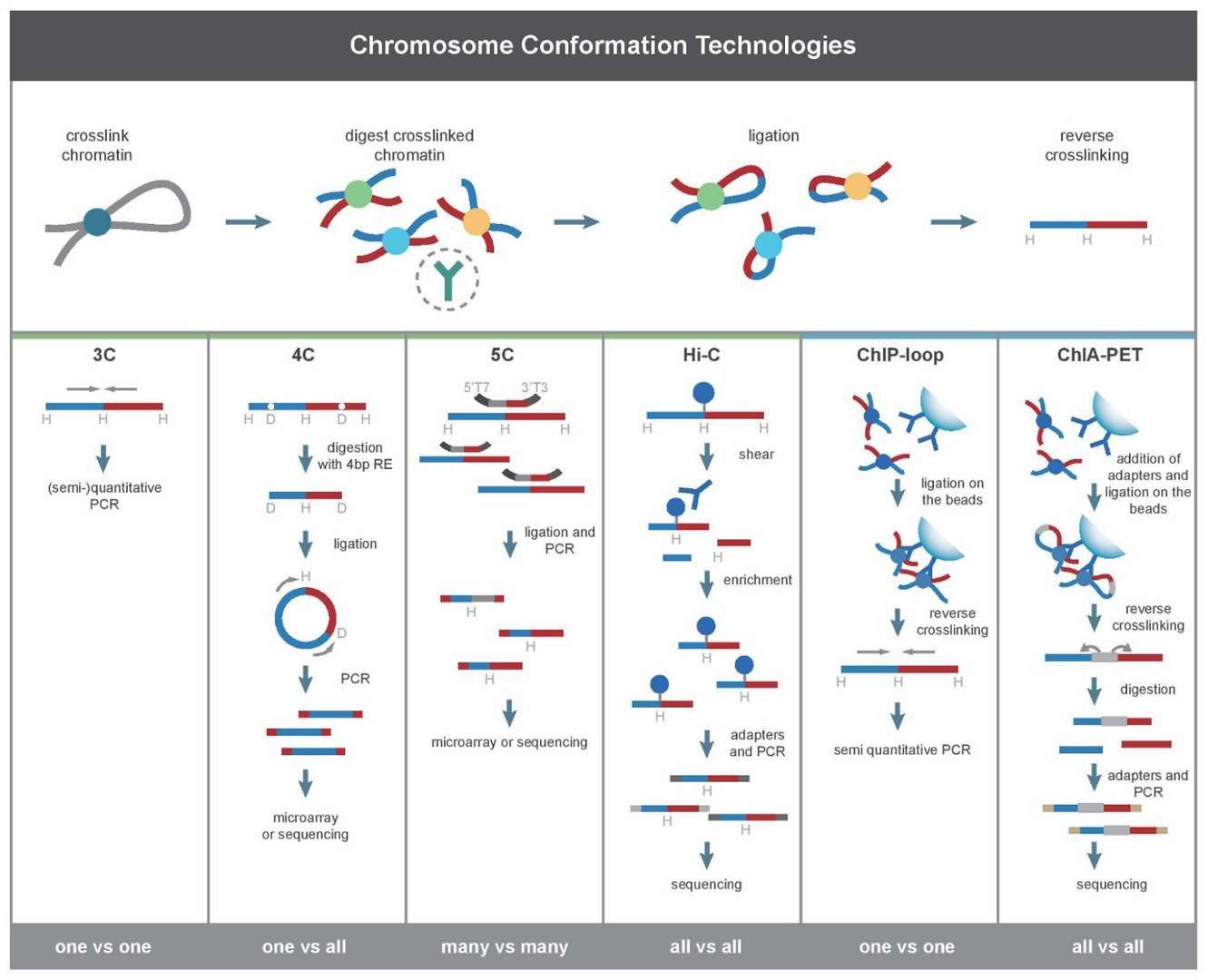 Development of Hi-C Technology (Li et al., 2014)
Development of Hi-C Technology (Li et al., 2014)
Service you may intersted in
Learn More:
The process of sample preparation for Hi-C experiments is of paramount importance to the quality of the resulting data. The primary challenge lies in the accurate capture of in situ three-dimensional interactions of chromosomes while circumventing technical biases introduced by human factors. This process necessitates a delicate balance between the meticulous operations of molecular biology and the stringent preservation of cellular states. Each stage, from cell fixation to DNA ligation, must be meticulously adjusted within a standardized procedure to address the distinct requirements of diverse sample types. In the following exposition, we will initiate our discussion with a standardized implementation of the experimental steps and the key strategy of quality control. We will then proceed to systematically explicate the efficient preparation of Hi-C samples and the assurance of data reliability.
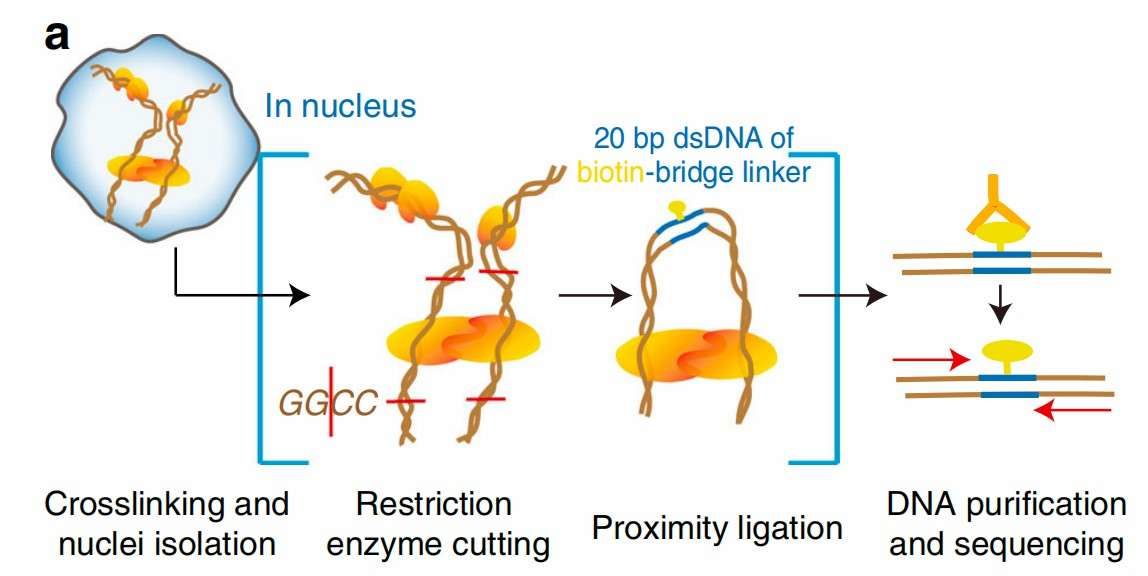 Workflow of Hi-C (Liang et al., 2017)
Workflow of Hi-C (Liang et al., 2017)
Detailed steps for preparing samples
The preparation of Hi-C samples commences with the chemical cross-linking of cells, marking the initial phase of "freezing" the spatial conformation of the chromatin. To immobilize the physical proximity of the chromatin, live cells are typically treated with a 1% formaldehyde solution for a duration of 10 minutes, facilitating the cross-linking of formaldehyde molecules with DNA-protein complexes. For samples requiring special handling, such as plant cells or fungi, where the presence of the cell wall may impede the penetration of the cross-linking agent, DSG, a membrane-penetrating cross-linking agent, can be used for pretreatment for 15 minutes before overlaying formaldehyde cross-linking to enhance the immobilization effect. Following the completion of cross-linking, it is imperative to immediately terminate the reaction by the addition of glycine (final concentration 0.25 M). The residual reagent must then be removed by centrifugation (500 × g, 5 min). The timeliness of this step directly determines the risk of over- or under-cross-linking.
Subsequent lysis of the cell to release chromatin constitutes a critical turning point in sample processing. Cell and nuclear membranes are disrupted using lysis buffers containing detergents (e.g., NP-40 or Triton X-100), and protease inhibitors (e.g., PMSF) are added to prevent degradation of DNA by endogenous nucleases. The lysed chromatin are then digested with restriction endonucleases to cleave the genomic DNA into linkable fragments. The digestion strategy must be customized according to the research objectives. For instance, MboI (recognition site GATC) is suitable for high-resolution studies due to its dense distribution in the genome, whereas HindIII (recognition site AAGCTT) is more suitable for genome-wide interactions mapping. The efficiency of enzymatic cleavage can be verified by pulsed-field gel electrophoresis (PFGE). The presence of DNA fragments with a size range of 1-10kb indicates sufficient enzymatic cleavage, whereas the occurrence of high molecular weight trailing necessitates either prolongation of the enzymatic cleavage time or adjustment of the Mg2+ concentration in the buffer.
The repair of enzymatically cleaved DNA ends necessitates biotin labeling to establish a reaction base for subsequent neighbor joining. The introduction of sticky ends was accomplished through the use of Klenow fragments, and A tails were incorporated in the presence of dATP to enable ligation to the T/A cloning junction. Subsequently, the ligation reaction of the spatially neighboring fragments was catalyzed using T4 DNA ligase in a highly diluted DNA solution (1 ng/μL). This step necessitates strict temperature control (16°C, 4 h) to avoid fluctuations in enzyme activity, as well as gentle mixing (e.g., rotary incubation) to ensure reaction homogeneity. The ligation products are enriched for biotin-labeled DNA fragments by streptavidin magnetic beads, a process that effectively removes unlinked free DNA and increases the percentage of target sequences.
Tips for ensuring high-quality samples
Quality control of Hi-C samples is carried out throughout the experiment, with a focus on minimizing the interference of DNA degradation and non-specific ligation. First, precise control of the cross-linking time is imperative: excessive cross-linking (>15 min) may lead to excessive chromatin condensation, impeding effective cleavage by restriction enzymes, while insufficient cross-linking (<5 min) may result in the dissociation of chromatin structures during subsequent manipulations, leading to the loss of interaction signals. To ascertain the optimal cross-linking time, it is recommended to conduct a preliminary experiment. In this experiment, a small sample is taken after cross-linking and sonicated. If the DNA fragment size is concentrated at 300-500 bp (as detected by an Agilent Bioanalyzer), this indicates moderate cross-linking.
Secondly, the restriction digest step necessitates meticulous observation of environmental interferences. For instance, residual SDS (used to facilitate chromatin depolymerization) may impede the enzyme activity, necessitating the reduction of SDS concentration through centrifugation or dilution prior to digestion. Furthermore, the lipid-rich chromatin of certain cell types (e.g., neurons or adipocytes) may adsorb restriction enzymes, thereby reducing their efficiency. This can be mitigated by adding bovine serum albumin (BSA, final concentration 0.1 mg/mL) to stabilize the enzymes. For clinical samples (e.g., FFPE tissues), an additional DNA repair treatment is required. These samples should be incubated overnight at 65°C with pre-repair buffer (containing proteinase K and RNase A) to reverse formaldehyde-induced DNA-protein cross-links and remove co-precipitated RNA impurities.
Finally, the specificity of the biotin labeling and ligation reaction must be verified in multiple dimensions. The presence of a junction dimerization peak after library construction (as indicated by a 125 bp peak on the Agilent Bioanalyzer) may suggest that the junction is overloaded in the ligation reaction. In such cases, the ratio of the junction to the DNA fragments must be adjusted (typically 1:10). Furthermore, batch-to-batch variations in streptavidin magnetic beads may impact the enrichment efficiency. Therefore, it is advisable to test each batch of magnetic beads for recovery by standard DNA (e.g., biotin-labeled λ DNA) prior to use, in order to ascertain the stability of their binding ability. By implementing these strategies, researchers can achieve a substantial enhancement in the signal-to-noise ratio of Hi-C data, thereby establishing a robust foundation for subsequent 3D genome reconstruction.
Hi-C sequencing represents the core stage of transforming chromatin spatial interactions into analyzable DNA sequence data. The technical challenge lies in transforming complex spatial junction fragments into high-fidelity and high-coverage sequencing libraries. This process requires precise molecular manipulation to transform DNA ligation products into sequencing-compatible libraries. It also requires the selection of appropriate sequencing strategies to balance data throughput and resolution according to research objectives. The accuracy of 3D genome reconstruction is contingent on the standardized operation and quality control of each step, from the enzymatic reaction of library construction to the optical signal acquisition of the sequencer. The following discussion will address the systematic operation of the sequencing process and the analysis of typical problems in the data generation process. This discussion will illustrate how to realize the efficient acquisition and accurate interpretation of Hi-C data.
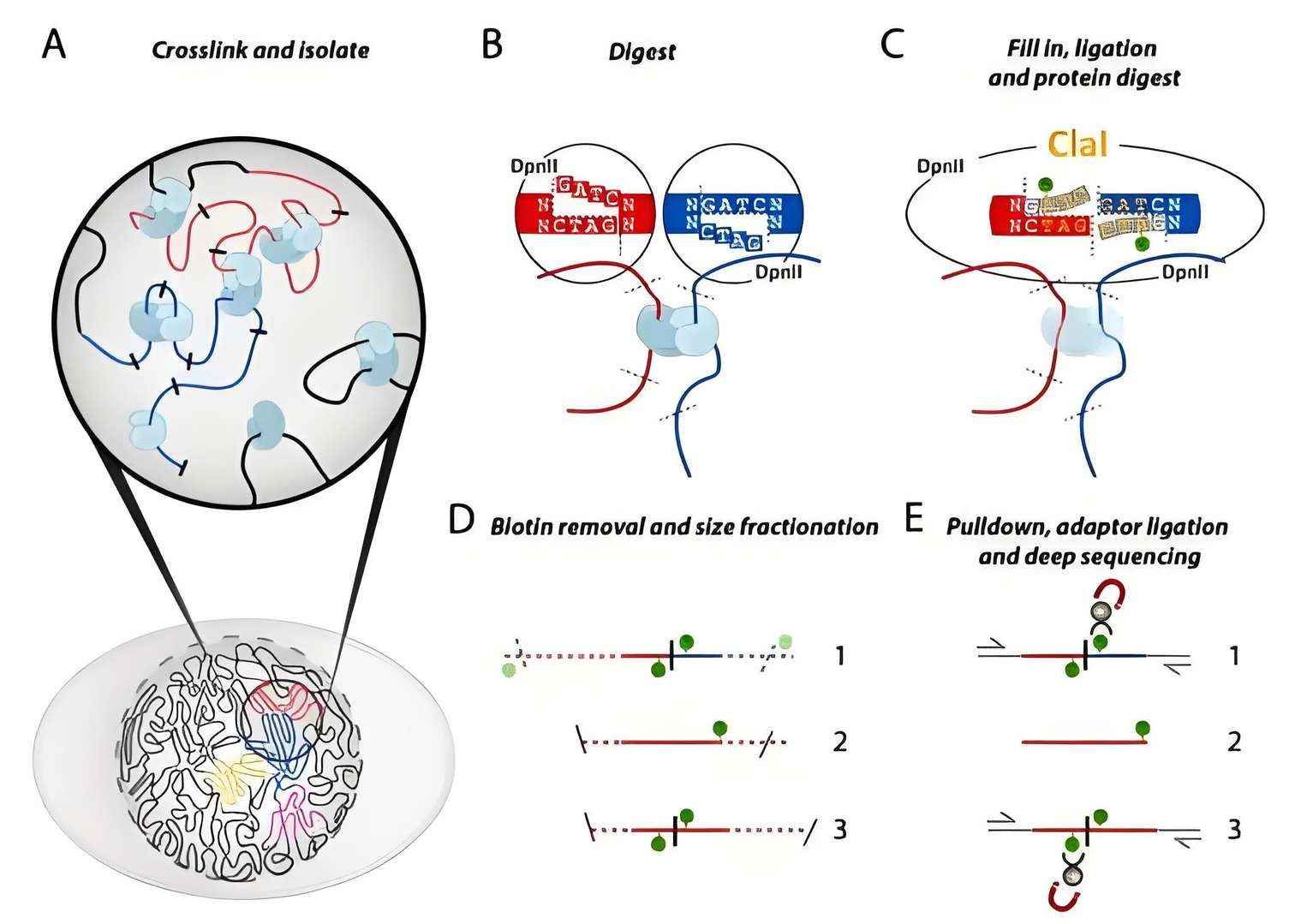 Schematic diagram of Hi-C technology
Schematic diagram of Hi-C technology
Step-by-step guide to the sequencing process
The construction of the Hi-C sequencing library commenced with the end repair of the enzymatic ligation products and junction joining. Initially, DNA fragments underwent end-repair using T4 DNA polymerase and T4 polynucleotide kinase to flatten sticky ends and phosphorylate the 5' end, thereby generating a flat-end structure for subsequent junction joining. Subsequently, an A-tail is added to the repaired DNA ends by Klenow fragmentation (3'→5' exo-) to enable specific ligation to sequencing junctions with a T protruding end. This step requires a tightly controlled reaction time (typically 30 minutes at 37°C) to avoid the formation of a junction dimer due to an excessively long A-tail. The design of the junction should include a Unique Dual Index (UDI) to support multiplex sequencing and a biotin-labeled capture sequence for subsequent magnetic bead enrichment.
Library amplification is a critical step in enhancing the signal strength of sequencing. PCR amplification with a limited number of cycles (typically 6-12 cycles) is performed using a high-fidelity DNA polymerase (e.g., Phusion or KAPA HiFi) to amplify DNA fragments with junctions. Subsequent to this, the amplified libraries are purified by magnetic beads (e.g., AMPure XP) to remove short fragments (<300 bp) and residual primers. Then, the library fragments are quantitatively assessed for library fragment size and concentration by Agilent Bioanalyzer or Qubit. For high-complexity samples, such as mammalian genomes, it is recommended that the main peak of the library fragments be located in the 400-700 bp range to ensure that the sequencing read lengths match the insert fragments.
The selection of a sequencing platform is contingent upon a judicious evaluation of factors such as the desired resolution and the cost-effectiveness of the study. In the context of genome-wide Hi-C studies, the Illumina NovaSeq 6000 platform has emerged as the preferred option due to its substantial throughput capacity (up to 3 Tb of data per flow cell) and its inherent advantage in terms of the read length of 150 bases. Conversely, PacBio HiFi or Oxford Nanopore long-read length sequencing is more suitable for haplotype resolution of repetitive regions, such as telomeres/mitochores. The sequencing depth is dynamically adjusted according to genome complexity. For human samples, 1-2 billion active reads are typically required to achieve 1 kb resolution, whereas microbial genomes can be sequenced with the same precision using 100 million reads.
Common issues and troubleshooting
The quality of Hi-C sequencing data is susceptible to multiple confounding factors during library preparation and sequencing. Among them, PCR amplification bias is the main causative factor for library complexity reduction. If the number of PCR cycles is too high (>15 cycles), it may lead to preferential amplification of high GC content fragments or short fragments, resulting in uneven genome coverage. This problem can be mitigated by pre-experimentation to optimize the number of cycles (with the lowest number of cycles to reach the library concentration threshold) or by adopting enzymatic ligation-conjugation alternatives (e.g., Tn5 transposase-mediated tagmentation). Furthermore, junction dimer contamination, evident as the 125bp peak in the Bioanalyzer electropherogram, is typically caused by excess junctions or incomplete purification. This can be addressed by adjusting the junction-to-DNA molar ratio (1:5 to 1:10 is recommended) or by increasing the number of magnetic bead purifications.
In the context of multiplex sequencing, two significant risks warrant particular attention: sample cross-contamination and index hopping. The utilization of UDI in multiplex sequencing entails a specific set of considerations. In instances where the molar concentration difference between samples is substantial, resulting from inaccurate library quantification, there is a possibility that the indexes of low-concentration samples may be erroneously labeled by the free junctions present in the high-concentration samples. To mitigate these risks, it is strongly recommended to employ accurate quantification of the library by qPCR and to ensure that the molar concentration difference between samples within the same sequencing lane does not exceed 20%. In the context of clinical samples, the presence of exogenous microbial or host DNA contamination poses a significant challenge, as it can obscure the target signal. This issue can be addressed through bioinformatics filtering, such as the elimination of unmatched reads following reference genome matching, or by implementing micro-sorting techniques prior to experimentation.
Low-quality reads (Q30 < 80%) in sequencing data may result from library degradation or abnormalities in the sequencer optical system. If FastQC reports a significant degradation of read quality at the 3' end of the reads, it is necessary to check whether the library fragment size meets the upper limit of the sequencing read length (e.g., PE150 mode of NovaSeq 6000 requires library fragments >350 bp). Low cluster densities or phase shifts (phasing) that occur during sequencing can be rectified by recalibrating the sequencer optics or optimizing the library upconcentration (typically 1.8-2.2 nM).With the above systematic nature control and error correction strategies, the reliability of Hi-C data and the biological significance of 3D genome models can be significantly improved.
Hi-C technology, a core tool for 3D genomic research, has profoundly reshaped our knowledge of chromatin spatial organization and functional regulation. In this study, we systematically analyzed the key links in the sample preparation and sequencing process, and confirmed the decisive role of standardized experimental operations and dynamic quality control strategies on data fidelity. In the initial stage of sample preparation, the spatial and temporal precision of chemical cross-linking, in conjunction with the specificity control of restriction enzyme cleavage, effectively preserves the in situ interactions of chromatin. In the subsequent stage of sequencing, the optimization of library construction based on enzymatic modification, in conjunction with the selective adaptation of sequencing platforms, significantly enhances the resolution of complex genomic regions. These technological breakthroughs have not only provided high-resolution research tools for revealing three-dimensional genomic aberrations and chromosomal rearrangements in cancer, but also promoted the discovery of epigenetic therapeutic targets.
However, the clinical translation of Hi-C technology is still facing significant challenges. First, the technology's sensitivity to low starting volume samples, such as circulating tumor DNA, is inadequate. Second, the high cost of long-read and long-sequencing limits its large-scale application. Third, the ability of data integration algorithms to resolve chromatin dynamics needs to be improved. To that end, future research should prioritize the following: microfluidic integration of single-cell Hi-C technology, CRISPR-mediated targeted chromatin capture, and other cutting-edge approaches. Additionally, the development of a cross-omics joint analysis framework is essential for realizing the multi-dimensional correlation analysis of 3D genome structure with transcriptional regulation and epigenetic modification. Through continuous technology iteration and interdisciplinary collaboration, Hi-C technology is expected to open up broader application frontiers in the fields of precision medicine and synthetic biology.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.