While substantial genomic advancements have transformed the landscape of scientific exploration, our knowledge regarding cis-regulatory elements in the pig genome remains notably limited. This knowledge gap significantly hinders the genetic enhancement and productivity of pigs, both as a meat source and as biomedical research models. In this investigation, the authors conducted a comprehensive examination of the genomes of four distinct swine breeds, employing a diverse array of cutting-edge histological techniques, including RNA-seq, ATAC-seq, ChIP-seq, and Hi-C. The study's strategy is reminiscent of large-scale epigenetic initiatives such as ENCODE (Encyclopedia of DNA Elements) and the Roadmap Epigenomics projects.
Cis-regulatory elements and their functions were systematically delineated in a dozen different tissues across these four pig breeds. This research generated a substantial dataset of 199 epigenetic regulatory profiles, culminating in the identification of over 220,000 cis-regulatory elements within the pig genome. Interestingly, this exploration revealed an unexpected level of conservation in cis-regulatory elements between the human and pig genomes, surpassing the conservation seen between the human and mouse genomes.
Furthermore, the research exposed variations in the structural domains associated with topological features within the porcine and human genomes, shedding light on the evolutionary changes impacting craniofacial morphology. Beyond its significance for porcine functional genomics and trait regulation, this study supplies essential comparative epigenetic data, enhancing the utility of pigs as models in human biomedical research.
Cis-regulatory Elements and 3D Genomic Structure of the Pig Genome
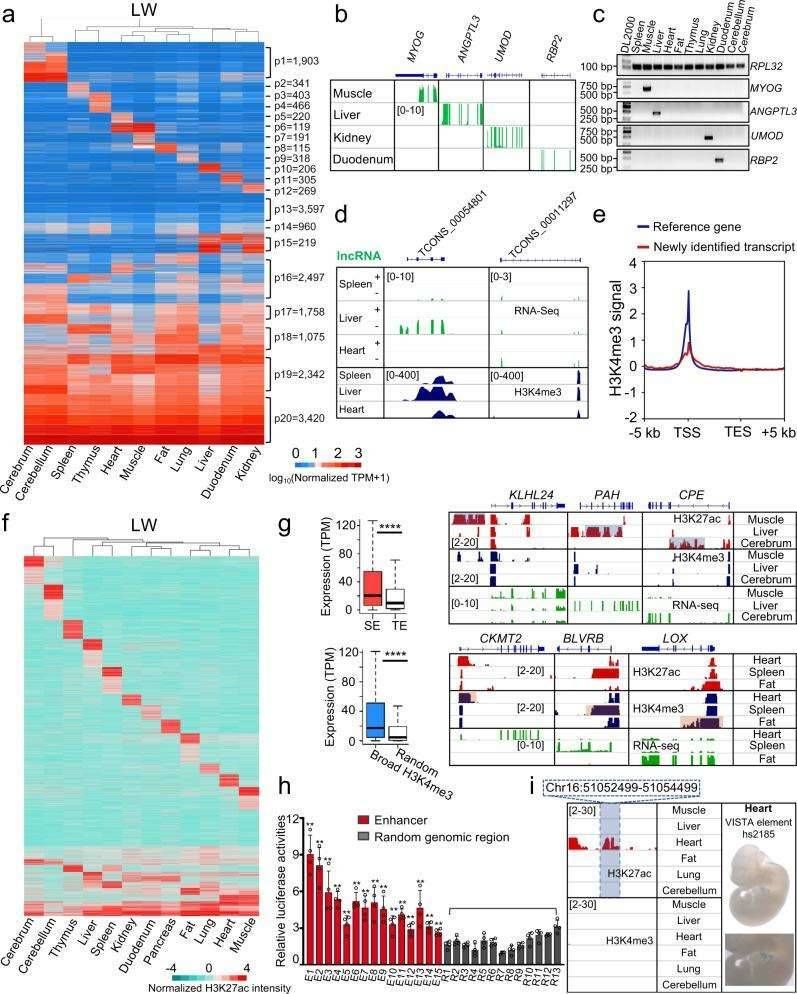
They found a total of 220,723 non-redundant cis-regulatory sequences, including 37,838 putative promoters, 146,399 potential enhancers, and 137,838 open chromatin regions aligned to the susScr11 genome. They examined the distribution of ChIP-seq and ATAC-seq signals around their TSSs and assessed their transcriptional expression levels. As examples, they showcase the AGL and FRRS1 genes on porcine Chr4 and the MYOG gene on Chr9.
The total length of these non-redundant cis-regulatory sequences is about 434.92 million base pairs, which is about 17.38% of the susScr11 genome. To assess the localization accuracy of the above-identified cis-regulatory sequences, they compared the enhancers and promoters with the TSS annotated by the University of California, Santa Cruz (UCSC) Swine Project and with previously published ChIP-seq data from porcine pluripotent stem cells and liver tissues. The results indicate that approximately 50% of the putative promoters are consistent with promoter overlaps or TSSs identified in the published data, while the other approximately 50% are not reported in the porcine genome. More than 86% of the enhancers were also not reported in the pig genome.
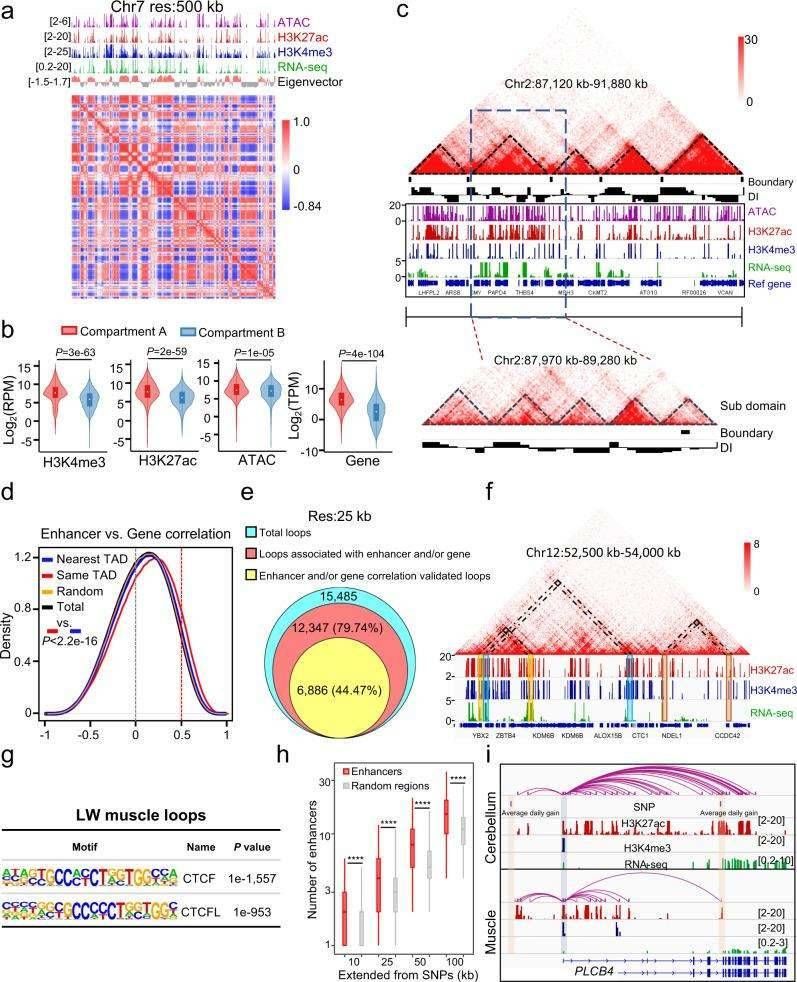
The 3D structure of the porcine genome was assessed using in situ Hi-C data, with skeletal muscle from a representative LW pig tissue sample. In total, 1,189,583,975 paired-end reads were sequenced, achieving more than 21x genome coverage, and they obtained 408,546,465 uniquely valid contacts, of which 290,325,259 were cis-contacts, after filtering for valid data using Hi-C-Pro. From these contacts, they mapped chromatin conformations as chromatin interaction frequencies, and the 3D genome structure modeling clearly showed the spatial relationships among pig genomic regions.
 Cis-regulatory element landscape of the pig genome. (Zhao et al., 2021)
Cis-regulatory element landscape of the pig genome. (Zhao et al., 2021)
Transcriptome of the Pig Genome
In their study, they conducted RNA-seq analysis on 52 samples derived from 11 different pig tissues across four distinct pig breeds. They revealed diverse patterns of RNA expression within each tissue, which they subsequently classified into 20 distinct clusters using the K-means function in the R programming language.
Cluster p20 stood out as a cluster with genes highly expressed in all samples. Further analysis, utilizing DAVID Gene Ontology (GO) enrichment, indicated that the genes in this cluster primarily play essential roles in fundamental biological processes, suggesting they may be considered housekeeping genes. Notably, more than half of the clusters exhibited a clear trend of tissue-specific expression.
They went on to identify 4,510 tissue-specific genes, defined as those showing at least a threefold higher expression in a particular tissue compared to others across all pig breeds. Subsequent DAVID GO enrichment analyses illustrated that they were significantly enriched for specific functions in a variety of tissues. They validated their findings by examining typical examples, and the results revealed a high level of agreement between RNA-seq data and RT-PCR results, reinforcing the accuracy of their analysis.
Moreover, they identified 3,316 novel transcripts, which included 1,713 long non-coding RNAs (lncRNAs) not previously documented in the porcine transcriptome. Strikingly, similar numbers of novel transcripts were detected in all examined tissues, suggesting that prior studies may have overlooked these specific transcripts. Notably, they found abundant H3K4me3 signaling near the transcription start site (TSS) of these newly identified transcripts, providing strong evidence for their active transcription. This robust identification process underscores the advantages of constructing strand-specific libraries after removing ribosomal RNA (rRNA), a technique rarely employed in previous porcine studies. Additionally, their results indicate that these newly identified transcripts exhibit a higher tissue-specificity index when compared to genes already annotated in the genome.
Transcriptional profiling and cis-regulatory elements analysis. (Zhao et al., 2021)
Tissue-Specific Enhancers: Insights into Porcine Gene Expression Patterns
Enhancer sequences serve as pivotal regulatory elements governing tissue-specific gene expression, exerting profound functional impacts on the establishment of distinct gene expression patterns. In this study, the authors meticulously categorized tissue-specific patterns associated with putative enhancers across various porcine tissues, successfully pinpointing 15,753 tissue-specific enhancers with a high level of confidence. Additionally, employing the ROSE algorithm, they also uncovered 414-1,306 super-enhancers in each tissue for each breed. As anticipated, genes associated with these super-enhancers exhibited markedly elevated expression levels in comparison to genes linked to typical enhancers.
Widespread H3K4me3 peaks, in conjunction with active promoters rich in H3K27ac, have been previously documented to significantly drive enhanced transcriptional activation of genes. The findings from this investigation revealed the presence of 418-1899 wide H3K4me3 peaks in each tissue across various breeds. Similar to the data on super-enhancers, genes proximal to these wide H3K4me3 peaks exhibited notably heightened expression levels compared to randomly selected genes.
To validate the robustness and precision of their methodology, the researchers conducted a dual luciferase reporter gene assay in porcine 3D4/21 cells, targeting 15 predicted non-tissue-specific enhancers and 18 promoter sequences chosen at random. The results underscored a substantial increase in transcriptional activity for the tested enhancers and promoters in comparison to randomized genomic regions. It's also worth noting that among the identified enhancers, 1216 sequences displayed conservation with known human VISTA enhancers.
 3D structure and regulation of cis-regulatory elements. (Zhao et al., 2021)
3D structure and regulation of cis-regulatory elements. (Zhao et al., 2021)
3D Genomic Structure of the Pig Genome
The identification of chromatin loops was facilitated through Hi-C matrix analysis. Using the enhanced HiCCUPS algorithm, we discovered 15,485 loops at a 25-kb resolution and 11,838 loops at a 40-kb resolution. A comprehensive amalgamation of Hi-C and cis-regulatory element data highlighted that, at the 25-kb resolution, 79.74% (12,347) of these loops were associated with cis-regulatory elements, with 44.47% exhibiting significant associations. Subsequent analyses, integrating loop data with open chromatin regions identified by ATAC-seq, demonstrated a substantial enrichment of CTCF-binding motifs within the loop anchors. These findings underscore the conserved role of CTCF-binding domains in shaping the 3D structure of mammalian genomes.
To explore the global impact of enhancers on the regulation of complex traits in pigs, the authors gathered SNPs that exhibited significant associations with published genome-wide association studies (GWAS) and examined their proximity to enhancers. A total of 7,238 GWAS-associated SNPs, of which 3,445 were non-redundant, were collected. The analysis revealed a notable enrichment of enhancers around SNPs significantly associated with GWAS in comparison to random genomic regions at varying distances. Notably, previous research had implicated the PLCB4 gene as a candidate gene for growth and average daily gain in pigs, and our study confirmed that the SNP significantly linked to the daily gain of pigs is located near an enhancer significantly associated with the PLCB4 gene.
Reference:
- Zhao, Yunxia, et al. "A compendium and comparative epigenomics analysis of cis-regulatory elements in the pig genome." Nature communications 12.1 (2021): 2217.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


