
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
In the field of biological science, the exploration of gene function is always one of the core tasks. Scientists have been trying to reveal the complex relationship between genes and life phenomena, and the emergence of CRISPR (clustered regularly spaced short palindromic repeats) technology has brought revolutionary changes to this exploration. It opens a new era of accurate gene editing and allows researchers to go deep into the microscopic world of genome. As a powerful research method of functional genomics based on CRISPR technology, CRISPR screening can systematically analyze gene functions at the whole genome level. Its core principle is to construct a library containing a large number of gRNA(guide RNA), which acts as an accurate "navigator" to guide gene editing tools to specifically edit many genes in a cell population. By observing and analyzing the edited cell phenotype, researchers can gradually reveal the relationship between genes and phenotypes, and then discover the key genes in specific biological processes. The following will introduce each key link of CRISPR screen technical process in detail.
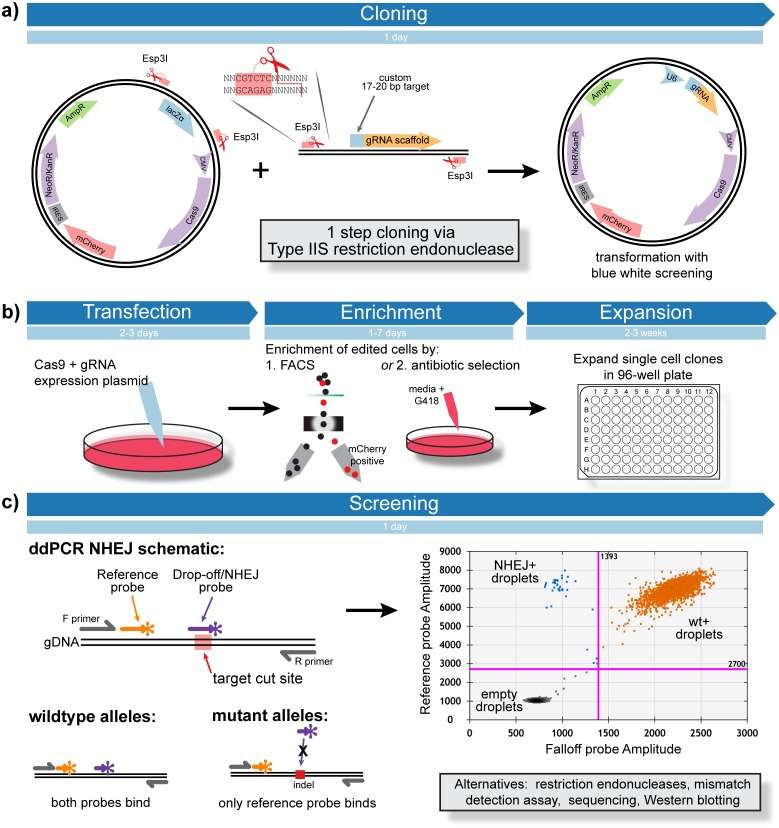 Overview of CRISPR Screening pipeline (Findlay et al., 2016)
Overview of CRISPR Screening pipeline (Findlay et al., 2016)
CRISPR-Cas system is a variety of gene editing "toolbox", among which CRISPR-Cas9 and CRISPR-Cas12 systems are the most widely used. CRISPR-Cas9 system consists of Cas9 protein and gRNA. GRNA recognizes the target gene sequence according to the principle of base complementary pairing, while Cas9 protein is like "molecular scissors" to cut DNA double-stranded at a specific position. The DNA repair mechanism of cells may lead to gene knockout when repairing the cleavage site. If foreign DNA fragments are introduced at the same time, gene insertion or replacement can be realized. In plant genetic engineering, researchers use this system to knock out the genes that regulate disease susceptibility in rice, thus enhancing the disease resistance of rice.
CRISPR-Cas12 system can not only cut double-stranded DNA, but also target single-stranded DNA under certain conditions. This feature has been applied in virus detection research, and the research team has developed a highly sensitive virus nucleic acid detection method. When the system recognizes and binds to the target virus nucleic acid sequence, it will activate the activity of non-specific single-stranded nuclease to cut the surrounding single-stranded DNA. The researchers realized the rapid and sensitive detection of virus nucleic acid by designing fluorescent labeled single-stranded DNA reporter molecules.
Different experimental purposes require different gene editing tools. CRISPR-Cas9 system is usually the first choice to explore the effect of gene deletion on cell phenotype and achieve complete gene knockout. Its technology is mature, which can stably cut the key exons of gene coding region and make the gene function lose. In the study of tumor cells, the researchers used this system to knock out the potential tumor suppressor gene, and observed that the proliferation, migration and invasion of tumor cells were enhanced, indicating that the gene had an inhibitory effect on the growth and metastasis of tumor cells.
When the purpose of the experiment is to carry out accurate base editing, introduce or correct point mutation, the optimized CRISPR derivation technology such as base editor is more suitable. The base editor can directly modify specific bases without causing DNA double strand breaks, and realize accurate base conversion. In the treatment of hereditary diseases, aiming at thalassemia caused by single base mutation, researchers tried to repair the mutation site of pathogenic gene in hematopoietic stem cells of patients with base editor, which brought hope for curing this kind of diseases.
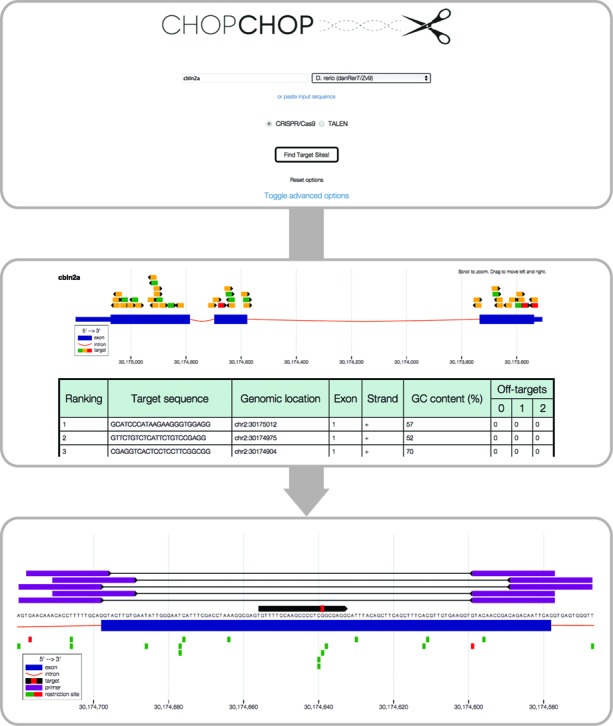 Workflow of a CHOPCHOP CRISPR/Cas9 query (Montague et al., 2014)
Workflow of a CHOPCHOP CRISPR/Cas9 query (Montague et al., 2014)
Take the Next Step: Explore Related Services
Learn More
Designing efficient and specific gRNA is the key initial step for the success of CRISPR screen experiment, and strict principles should be followed. GRNA targeting sequence must be highly specific to avoid base complementary pairing with non-target regions, so as to prevent off-target effect from affecting the experimental results. Researchers use bioinformatics algorithms to scan the genome and select unique sequences as targeting sites. For example, by comparing with the genome database, gRNA sequences with very low similarity to non-target sequences are retained.
The length of gRNA is generally between 18 and 23 bases, which is verified by a large number of experiments, which can not only ensure the effective combination with the target gene, but also maintain its own structural stability. At the same time, the GC content of gRNA sequence should be 40%-60%. Too high GC content is easy to form complex secondary structure, which hinders pairing with target sequence; If it is too low, the binding force is insufficient and it is easy to dissociate. When designing gRNA for specific genes, it is necessary to optimize GC content and improve editing efficiency.
Bioinformatics tools provide strong support for gRNA design and improve the efficiency and accuracy of design. There are many specialized softwares and online platforms on the market, such as CRISPOR and CHOPCHOP. These tools integrate a wealth of genome data and algorithms. When researchers input the target gene sequence, the tools can screen out potential gRNA sequences according to the design principles, and give parameters such as off-target risk prediction and potential editing efficiency. Taking CRISPOR as an example, it can design gRNA based on various CRISPR-Cas systems, and can also predict off-target sites in detail. Based on this information, researchers can select the gRNA sequence that is most suitable for the experiment.
Bioinformatics analysis of candidate sites for gene editing (such as QTL and GWAS) is very helpful for CRISPR screen. Narrowing the candidate editing sites to a certain range can greatly reduce the burden of the whole experiment. After choosing the right editing tool, the next step is to design gRNA library. Besides the editing efficiency, we must also carefully consider the off-target situation. At present, there are dozens of gRNA design websites available (Table 1). Some of these programs allow users to design multiple targets in the whole genome, such as CRISPy web and CRISPR Library Designer, which is very useful for obtaining the whole gRNA data set covering the candidate areas.
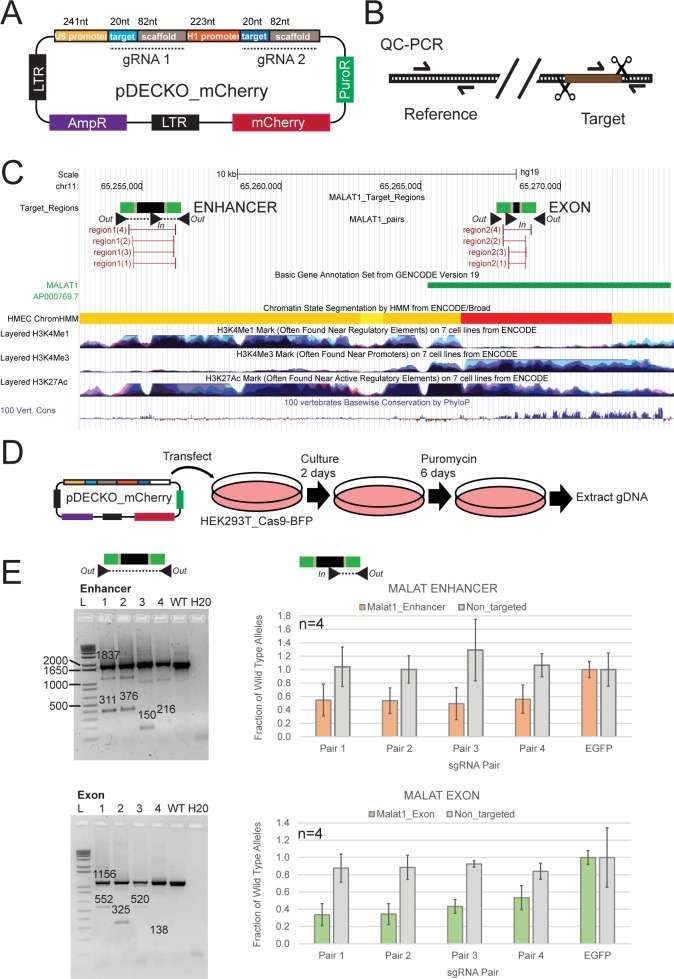 Measuring the deletion efficiency of paired sgRNA designs (Pulido-Quetglas et al., 2017)
Measuring the deletion efficiency of paired sgRNA designs (Pulido-Quetglas et al., 2017)
gRNA library mainly includes whole genome library and focused library. The whole genome library contains gRNA sequences designed for almost all genes in the genome, which is suitable for large-scale gene function screening of the whole genome and can comprehensively mine genes related to specific phenotypes. When studying the response mechanism of cells to new drugs, genes involved in drug metabolism, target and drug resistance can be found by screening the whole genome gRNA library. Focused libraries construct gRNA for specific gene families, signal pathways or functional categories. When researchers pay attention to the regulation mechanism of PI3K-Akt signaling pathway in tumor cells, constructing a library focusing on this pathway can reduce the workload and complexity of experiments and improve the efficiency of screening key genes.
The process of library construction is delicate and complex, and it needs multiple steps to cooperate closely. Firstly, a large number of gRNA oligonucleotide sequences were obtained by chemical synthesis or PCR amplification. Chemical synthesis can accurately control the sequence and has high purity; PCR amplification is suitable for large-scale amplification of known sequences. These oligonucleotide sequences are the basis of constructing the library. Then, the gRNA oligonucleotide sequence was cloned into suitable vectors, such as lentivirus vector, adeno-associated virus vector and so on. Take lentivirus vector as an example, it can efficiently infect many kinds of cells and stably integrate foreign genes. During cloning, restriction endonuclease and DNA ligase were used to cut out specific ends and connect them, so that the gRNA oligonucleotide sequence was inserted into the vector accurately and the recombinant vector was constructed. Subsequently, the recombinant vector was amplified and purified.
Usually, Escherichia coli is used to amplify the vector, cultured in a suitable medium to carry the recombinant vector, and then purified by alkali cracking and column chromatography to remove impurities and obtain high-quality library plasmids. Finally, the library plasmid was packaged into infectious virus particles by virus packaging system. The library plasmid and auxiliary plasmid were co-transfected into packaging cell lines (such as 293T cells), and complete virus particles were assembled in the cells for introducing gRNA library into cell population.
After the design of gRNA data set is completed, a large number of sgRNA will be cloned into CRISPR/Cas plasmid. It is particularly important to ensure the accuracy of cloning and the gRNA distribution of plasmid library. Negative selection marker ccdB can be used to improve the accuracy. The distribution of gRNA is maintained by using high coverage libraries (for example, > 30×). After careful evaluation by high-throughput sequencing (NGS), subsequent transformation was carried out.
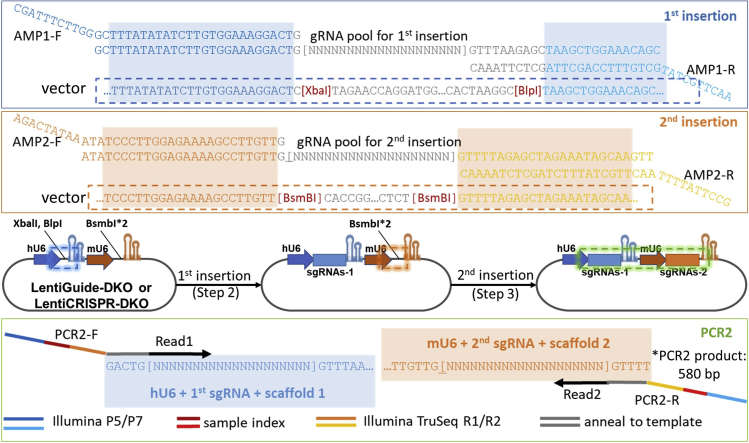 Overall library construction process and gRNA pools/primer design (Tang et al., 2022)
Overall library construction process and gRNA pools/primer design (Tang et al., 2022)
Selection of cell lines Choosing a suitable cell line is an important prerequisite for the success of large-scale genetic transformation, and many factors need to be considered comprehensively. Cell growth characteristics are very important. Cell lines that grow rapidly and are easy to culture (such as HeLa cell line) are beneficial to obtain a large number of cells and ensure the smooth progress of the experiment. The state of cell differentiation can not be ignored. When studying related topics of cell differentiation, we should choose cell lines with stable characteristics at a specific differentiation stage. When studying the differentiation mechanism of nerve cells, we can more accurately observe the influence of gene editing on the differentiation process by selecting cell lines with stable differentiation potential in neural stem cell stage. The sensitivity of cell lines to virus infection is also very important, because gRNA library needs to be introduced through virus vectors, and cell lines sensitive to virus infection can improve the introduction efficiency. 293T cell line is highly effective in infecting many viruses, and is often used in virus packaging and gene transduction experiments.
Transfection/infection method gRNA library was introduced into cell population mainly by transfection or infection. Transfection is suitable for cell lines that are insensitive to virus infection or easy to be physically transfected. The common methods are liposome transfection and electroporation transfection. Liposome transfection is to mix gRNA library plasmid with liposome to form a complex, and then the plasmid is sent to the cell after the liposome is fused with the cell membrane. Electroporation transfection uses electric pulses to punch holes in the cell membrane, so that the plasmid can enter the cell. By controlling the parameters of electric pulses, the introduction efficiency can be improved. For most cell lines, viral vector infection is a more efficient way of introduction. After packaging gRNA library into lentivirus or adeno-associated virus, virus particles can efficiently transfer the library into cells and integrate it into genome. The virus titer and multiplicity of infection (MOI) should be accurately controlled during infection. Through pre-experimental optimization, it is ensured that the cells can obtain an appropriate amount of gRNA to avoid over-infection or under-infection.
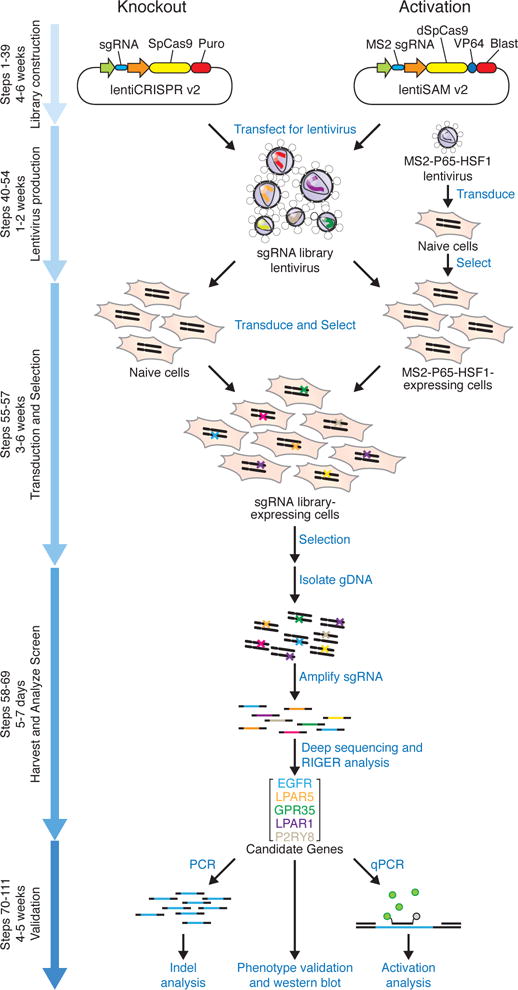 Transfection in CRISPR Screening (Joung et al., 2017)
Transfection in CRISPR Screening (Joung et al., 2017)
The key to genotyping is to accurately detect the changes of gene editing sites in cells, and there are many detection methods. PCR amplification combined with Sanger sequencing is a classic and common method. Firstly, specific primers were designed for the sequences on both sides of the editing site, and DNA fragments containing the editing site were amplified by PCR, and then Sanger sequencing was carried out. Comparing the sequencing results with the wild-type gene sequence, we can judge whether the gene editing is successful or not and the editing type. The result of this method is accurate and intuitive, but high-throughput sequencing technology has gradually become an important means. Targeted amplicon sequencing can simultaneously sequence multiple gene editing sites of a large number of samples in parallel. First, the target gene region is amplified to construct a sequencing library, and then high-throughput sequencing platform is used for sequencing. With the help of bioinformatics tools, a large number of accurate gene editing data can be quickly obtained, which improves the experimental efficiency.
Data analysis determines the situation of gene editing. After obtaining genotyping sequencing data, data analysis is the key to interpret gene editing information. First of all, we should control the quality of the original sequencing data, remove impurities such as low-quality sequencing reads and linker sequences, commonly use FastQC to evaluate the data quality, and trim and filter the low-quality data with Trimmomatic. Then the quality control data are compared with the reference genome by bioinformatics software, such as BWA and Bowtie. By comparing the sequence changes of identifiable editing sites, the frequency distribution of different editing types in the population is analyzed to evaluate the editing efficiency and effect. Comparing the differences of editing in different experimental groups can explore the relationship between gene editing and processing factors and provide clues for understanding gene function.
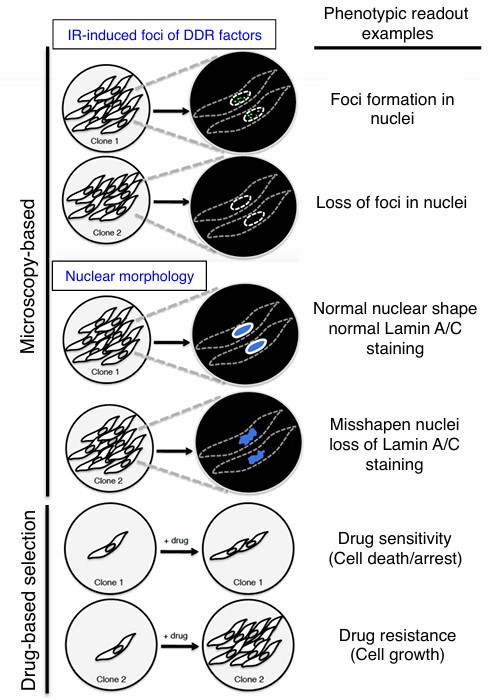 Genotyping in CRISPR Screening (Chiang et al., 2016)
Genotyping in CRISPR Screening (Chiang et al., 2016)
Learn More
The technical process of CRISPR Screening mainly includes: first, construct an sgRNA library, and design and synthesize the corresponding sgRNA sequence for the target gene. Then, the library was introduced into the cell population, so that sgRNA combined with Cas9 protein played a role in gene editing. Then, specific selection pressure is applied to the cells to screen out cells with specific phenotypes. After that, the genomic DNA of the cell was extracted and the region containing sgRNA was amplified. Finally, high-throughput sequencing was used to analyze the sequencing data, and the key genes affecting the phenotype were determined by comparing with the library sequence, thus revealing the gene function and related biological mechanisms.
References
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.


