- Home
- Solutions
- Epigenome Sequencing
- Metagenomics Sequencing
- DRUG-seq Service
In the field of biological science research, CRISPR screening and sequencing technology is setting off a research boom. CRISPR Screening data contains a lot of biological mysteries, which can accurately reveal gene functions, regulatory mechanisms and related signal pathways by high-throughput sequencing of specific gene editing libraries. These data are key clues, from exploring the pathogenesis of complex diseases to mining new drug targets. Interpreting this kind of data is like opening the door to the world of precise operation inside cells, which can help researchers break through the fog and bring unprecedented changes to basic research and clinical application of life sciences.
Interpretation of basic data is an important link in the analysis of CRISPR screening and sequencing results. Through the evaluation of data quality, editing sites, editing efficiency and off-target effect, we can preliminarily understand the effect of CRISPR editing in cells or organisms, and provide an important basis for subsequent functional research and application.
Sequencing depth refers to the average number of times each base in the genome has been sequenced. Sufficient sequencing depth is very important for accurate detection of gene editing events. Generally speaking, the higher the depth, the stronger the reliability of the data, and the more accurate the recognition of low-frequency editing events. If the sequencing depth is too low, some rare editing types may be missed, resulting in inaccurate results.
Base mass value is an index to measure the recognition accuracy of each base in the sequencing process. It is usually expressed by the mass fraction of Phred, and the higher the mass value, the greater the probability that the base is correctly recognized. High-quality base data is the basis of subsequent accurate analysis. If the base mass value is generally low, it will increase the possibility of mistaken base recognition and affect the judgment of editing sites and types.
Reference genome comparison: Compare the sequenced reads with the reference genome to determine the position of each read on the genome. By comparison, we can find different loci from the reference genome, which may be caused by CRISPR editing or natural genetic variation. Therefore, it is necessary to combine the experimental design with the control group to distinguish the real editing sites.
Indel detection: CRISPR editing often leads to insertion or deletion mutation at the target site. By analyzing the comparison results, Indel events near the editing site can be identified. The size and position of Indel are of great significance for understanding the editing mechanism and evaluating the editing effect. Smaller Indel (such as deletion or insertion of 1-several bases) may lead to frameshift mutation, thus losing gene function; However, larger Indel may cause more complicated changes in gene structure.
Frequency calculation: editing efficiency is usually expressed by the mutation frequency of editing sites. The editing frequency can be obtained by calculating the ratio of the number of reads with editing events at the target site to the total number of reads covering the site. The higher the editing frequency, the better the editing effect of CRISPR system at this site. Generally speaking, an efficient CRISPR editing system can produce a higher proportion of editing cells in the cell population, so it is easier to observe the changes of gene function.
Proportion of different editing types: CRISPR editing may produce many types of mutations, such as homozygous mutation, heterozygous mutation and biallelic editing. The complexity and diversity of editing events can be understood by analyzing the proportion of different editing types. For example, homozygous mutation may lead to complete loss of gene function, while heterozygous mutation may have different degrees of functional impact. Understanding the distribution of different editing types is helpful to evaluate the application potential of CRISPR editing in gene function research and gene therapy.
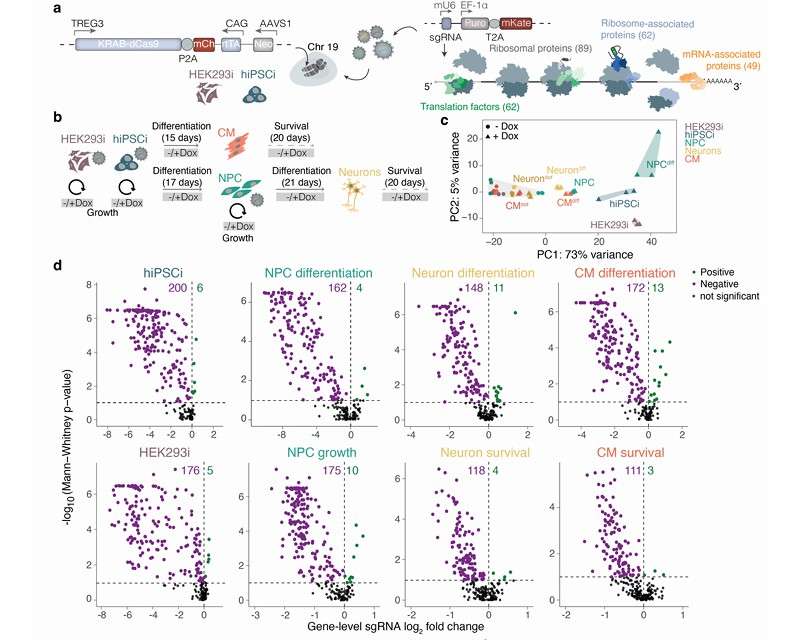 Comparative inducible CRISPRi screens identify essential components of the mRNA translation machinery in human cells (Rodschinka et al., 2025)
Comparative inducible CRISPRi screens identify essential components of the mRNA translation machinery in human cells (Rodschinka et al., 2025)
Take the Next Step: Explore Related Services
Learn More
The interpretation of CRISPR Screening-differential analysis results needs to comprehensively consider many aspects, combine biological knowledge and experimental background, and deeply explore the biological significance behind the data, thus providing valuable information for related research and application.
Differential gene list: The analysis results usually give a list of differentially expressed or differentially enriched genes. These genes show significant differences under different experimental conditions (such as control group and treatment group). Focus on those genes with large difference multiples and P value or adjusted P value (such as FDR, false detection rate) less than the set threshold (usually 0.05).
Functional annotation of genes: Functional annotation of differential genes to understand their biological processes, molecular functions and cell components. You can use databases such as Gene Ontology (GO), KEGG, etc. For example, it is found that the differential genes are mainly concentrated in cell cycle regulation and signal transduction pathways, suggesting that the corresponding biological processes may be affected in the experimental treatment.
Site information: If it is CRISPR screening for specific gene sites, we should pay attention to the position and type of different sites (such as base substitution, insertion, deletion, etc.). These sites may affect groups.
Enrichment analysis: Determine the signal pathway of significant enrichment of differential genes. Common path analysis methods include enrichment analysis based on hypergeometric distribution. For example, it is found that differential genes are enriched in PI3K-AKT signaling pathway, which indicates that this pathway may play an important role in the biological process targeted by CRISPR screening.
Path interaction: consider the interconnection between different paths and network regulation. One gene may participate in multiple pathways, and there are also complex upstream and downstream relationships and feedback regulation mechanisms between pathways. By constructing the pathway network, we can understand the synergy between genes and the overall regulation of biological systems more comprehensively.
Cell type marker genes: If the experiment involves multiple cell types, we should combine the cell type-specific marker genes to analyze the difference results. Determine which differential genes are unique to a specific cell type and the expression changes of these genes in different cell types. This is helpful to understand the difference of CRISPR screening in different cell types.
Change of cell type proportion: By analyzing the proportion change of different cell types in sequencing data, it is judged whether CRISPR screening has an impact on the composition of cell population. For example, the proportion of a certain cell type increased or decreased significantly in the treatment group, which may suggest that CRISPR screening has an effect on the proliferation, differentiation or survival of this cell type.
Discussion on biological mechanism: Based on the above analysis results, the biological mechanism revealed by CRISPR screening was deeply discussed. For example, clarifying the role of specific genes or pathways in disease occurrence, cell development, physiological processes, etc., provides theoretical basis for further understanding of life phenomena and disease mechanisms.
Discovery of therapeutic targets: If the research is related to diseases, the results of differential analysis may help to find potential therapeutic targets. According to these targets, we can further develop drugs or design treatment strategies, and provide new ideas and methods for the treatment of diseases.
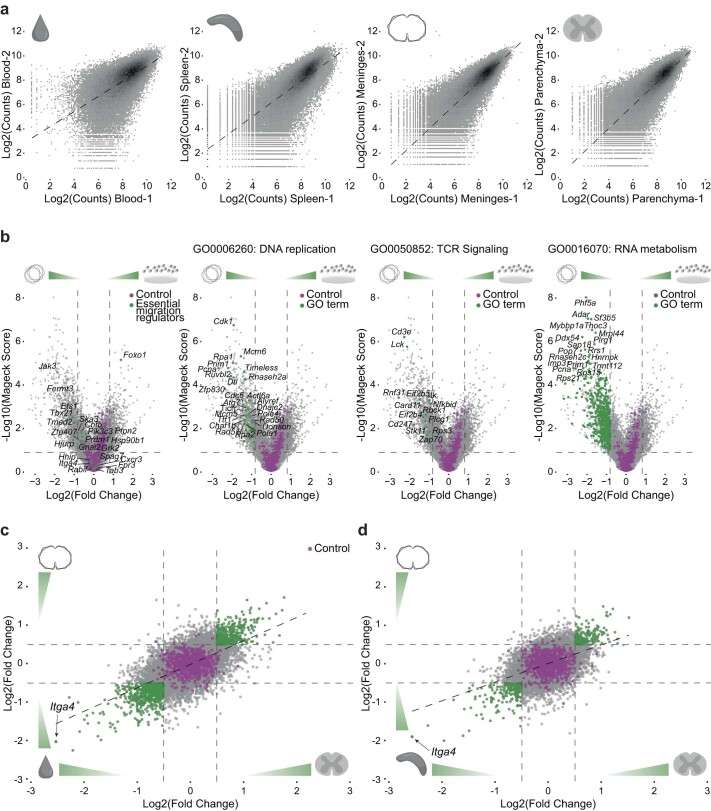 Analysis of the genome-wide CRISPR screen (Kendirli et al., 2023)
Analysis of the genome-wide CRISPR screen (Kendirli et al., 2023)
CRISPR Screening is a method of functional screening at the genome level by using CRISPR-Cas9 technology. By sequencing the screened cell population, the results at the gene level can be obtained. The following are some key points to interpret the results.
Sequencing quality evaluation: First, we should evaluate the quality of the original sequencing data, check the distribution of base quality values, sequencing error rate and other indicators to ensure the reliability of the data. Tools such as FastQC are usually used to accomplish this task. If the data quality is not good, it may need to be filtered and pruned to remove low-quality bases and linker sequences.
Alignment to the reference genome: The pretreated sequenced reads are aligned to the reference genome to determine the position of each read in the genome. Commonly used comparison tools include Bowtie, BWA and so on. The comparison results are usually saved in BAM or SAM file format, and these files contain the matching information between reads and genome, such as chromosome position, comparison score and so on.
Determine the insertion deletion sites: Based on the comparison results, find the insertion or deletion (Indel) sites in the genome, which are likely to be produced during the repair process after CRISPR-Cas9 cleavage. Special mutation detection tools, such as GATK and Samtools, can be used to identify these Indel mutations. Generally speaking, a small insertion deletion (usually less than 50 bases) is a common feature of CRISPR editing.
Verification of sgRNA specificity: Check whether the identified Indel site corresponds to the designed sgRNA targeting site. Ensure that editing occurs in the expected gene region and there is no obvious off-target effect. The specificity can be verified by looking at the complementary pairing between sgRNA and genome and analyzing the characteristics of surrounding sequences. If unexpected editing sites are found, it is necessary to further evaluate the possibility and influence of off-target effect.
Calculation of editing frequency: Evaluate the efficiency of gene editing by counting the proportion of cells editing at the target gene site. The editing frequency can be calculated according to the proportion of wild-type and editing alleles in sequencing data. For example, through deep sequencing, it is found that the frequency of editing alleles at a certain gene site is 30%, which means that the gene editing efficiency at this site is 30%.
Compare the efficiency of different sgRNAs: If multiple sgRNAs are used to target the same gene or different genes in the experiment, their editing efficiency can be compared to determine the optimal sgRNAs. sgRNA with high editing efficiency and good specificity is usually selected for follow-up research. sgRNA with high editing efficiency may have greater advantages in gene function research or gene therapy.
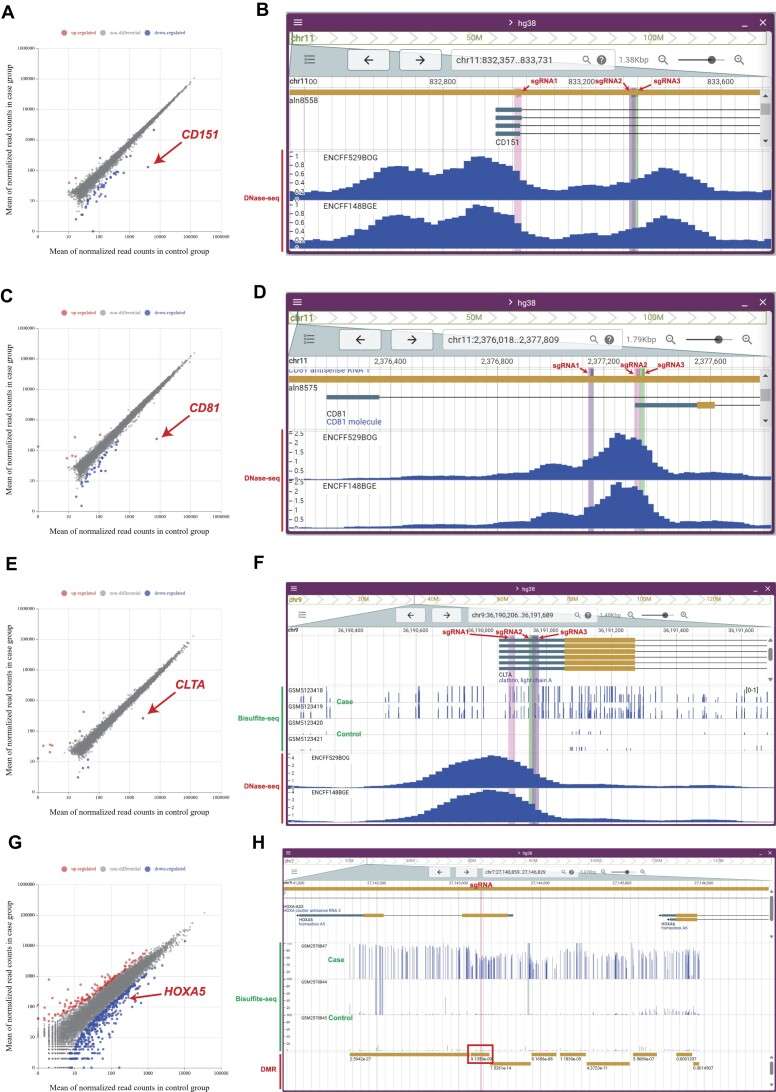 Case study of inhibition of gene expression in CRISPRepi (Shi et al., 2025)
Case study of inhibition of gene expression in CRISPRepi (Shi et al., 2025)
Enrichment analysis: If functional genomics research based on CRISPR screening is carried out, such as screening genes sensitive or resistant to certain drugs in cell population, gene enrichment analysis can be used to determine which genes or biological pathways have been significantly affected in the screening process. Commonly used enrichment analysis methods include GO(Gene Ontology) enrichment analysis and Kegg (Kyoto encyclopedia of genes and genomes) pathway enrichment analysis. These analyses can help to determine gene functions and biological processes related to specific phenotypes.
Phenotypic correlation analysis: The results of gene editing are correlated with the phenotypic changes of cells. For example, it is observed that after a gene is edited, the growth rate, apoptosis rate and metabolic characteristics of cells have changed, and the function of the gene can be inferred through this correlation. If gene editing leads to changes in the sensitivity of cells to a drug, then the gene may be related to the mechanism of drug action or the drug resistance of cells.
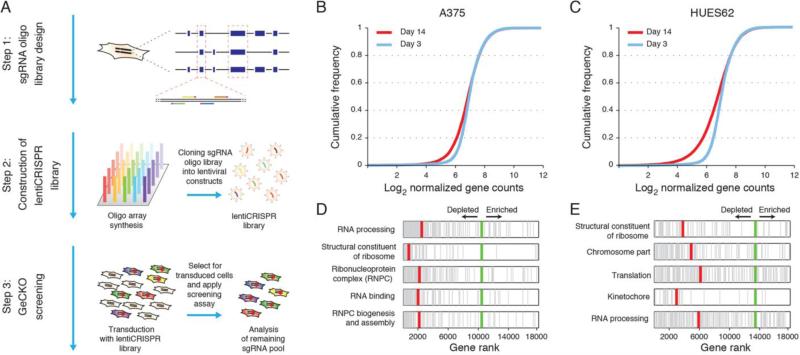 GeCKO library design and application for genome-scale negative selection screening (Shalem et al., 2014)
GeCKO library design and application for genome-scale negative selection screening (Shalem et al., 2014)
When interpreting the results of CRISPR screening and sequencing-gene level, it is necessary to comprehensively consider various aspects of information, combined with experimental design and biological background, in order to accurately infer the function of genes and the effect of CRISPR editing. At the same time, attention should be paid to the repeatability of the experiment and the reliability of the results, and further verification experiments should be carried out if necessary.
Integrating the results of CRISPR screening and sequencing with other data can help us understand the gene function and biological process more comprehensively. The following are some common integration methods and key points of interpretation.
The results of CRISPR screening were integrated with protein omics data such as protein expression level and protein modification. For example, the change of protein expression was obtained by mass spectrometry, or the modification such as protein phosphorylation was detected by antibody chip. Gene editing may affect the expression level of protein, post-translational modification or the stability of protein. If the editing of CRISPR leads to the decrease of protein expression of a gene, it may affect the gene transcription, mRNA stability or translation process.
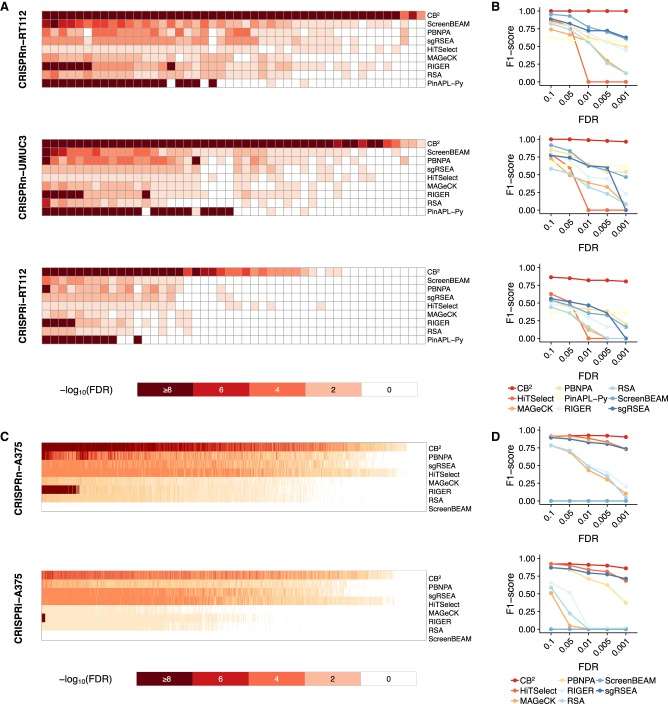 CB2 offers robust target identification with high precision and recall (Jeong et al., 2019)
CB2 offers robust target identification with high precision and recall (Jeong et al., 2019)
The results of CRISPR screening were correlated with the changes of metabolite levels. The types and contents of intracellular metabolites were detected by gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS). When a gene is edited by CRISPR, if the metabonomics data shows that the level of some metabolites has changed significantly, it means that the gene may be involved in the corresponding metabolic pathway.
In clinical research, the gene information obtained by CRISPR screening is combined with the clinical characteristics, treatment response and prognosis of patients. If it is found that the editing status of CRISPR of a gene is related to the disease stage, metastasis or treatment response to a specific drug, then the gene may be a key marker of disease progress or treatment response.
In data integration, we need to pay attention to data quality control, standardized processing and statistical analysis to ensure the accuracy and reliability of the results. At the same time, it is necessary to combine biological knowledge and research background to reasonably interpret and infer the integrated data, so as to mine more valuable information.
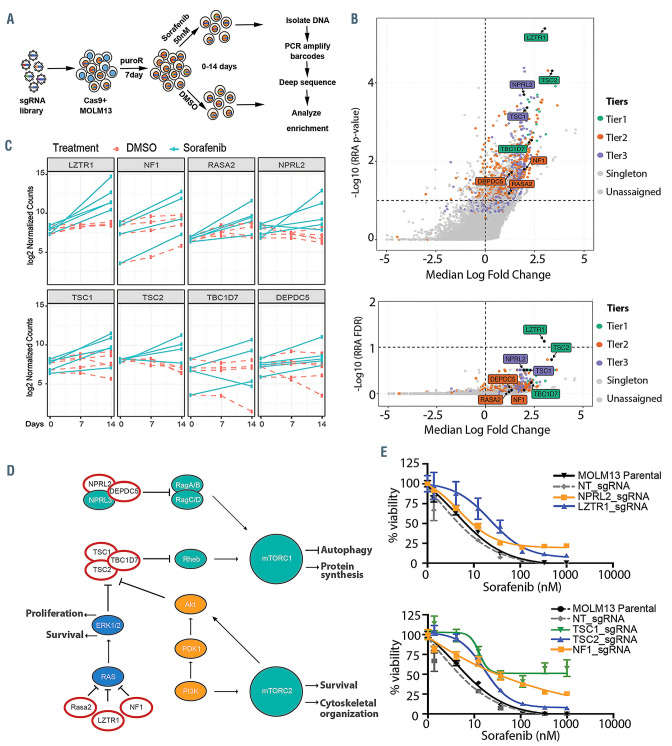 A genome-wide CRISPR knockout screen identifies negative regulators of MAPK and MTOR pathways (Damnernsawad et al., 2022)
A genome-wide CRISPR knockout screen identifies negative regulators of MAPK and MTOR pathways (Damnernsawad et al., 2022)
CRISPR Screening data can be used to analyze gene functions. By editing and sequencing the genome by CRISPR technology, genes related to specific phenotypes can be identified. When analyzing the data, the sequencing data should be controlled and compared first, and then the editing sites should be identified. The enriched sgRNA corresponding genes under positive selection may be the key to cell survival or phenotypic change; The corresponding gene of sgRNA deleted under negative selection may be an essential gene. With the help of these data, we can deeply understand the gene regulatory network, find potential drug targets, and help the study of disease mechanism and drug research and development.
References
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.



