
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
Forward genetic screening methods play a pivotal role in elucidating the molecular mechanisms underlying significant agricultural traits. These methods rely on genetic variations generated through natural mutations, physical and chemical mutagenesis, and T-DNA insertion. However, identifying genotypes with specific phenotypic characteristics using these methods often requires substantial time and effort. To address these challenges, researchers have developed large-scale screening methods based on RNA interference (RNAi). Despite their potential, these methods face limitations, including significant off-target effects and incomplete knockout phenotypes, compounded by stringent regulatory restrictions.
The CRISPR/Cas system, an advanced gene-editing tool, has emerged as a crucial driver in both fundamental research and crop breeding. Its efficiency, simplicity, and programmability have facilitated the rapid development and widespread application of CRISPR screening (whole-genome or high-throughput gene editing techniques) for large-scale editing. Remarkable progress has been made in mammalian studies using CRISPR screening. As the technology advances, it shows significant potential in crop research, with documented applications in various crops such as rice, tomato, soybean, and maize (Jacobs et al., 2017; Lu et al., 2017; Meng et al., 2017; Bai et al., 2020). It is anticipated that the number of gene-edited germplasms will significantly increase in the coming years with the broad adoption of this technology.
The development of high-quality, high-coverage, and uniformly distributed mutant libraries is crucial for functional genomics research and crop improvement. These libraries provide researchers with extensive genetic resources, fostering advancements in the field.
Take the Next Step: Explore Related Services
The CRISPR screening workflow encompasses several critical steps: selecting an appropriate gene-editing tool, designing a gRNA dataset, constructing a library, performing large-scale genetic transformation, genotyping, phenotypic characterization, and managing germplasm resources. This paper aims to provide a detailed overview of the CRISPR screening workflow and its practical applications, offering valuable insights and references for researchers.
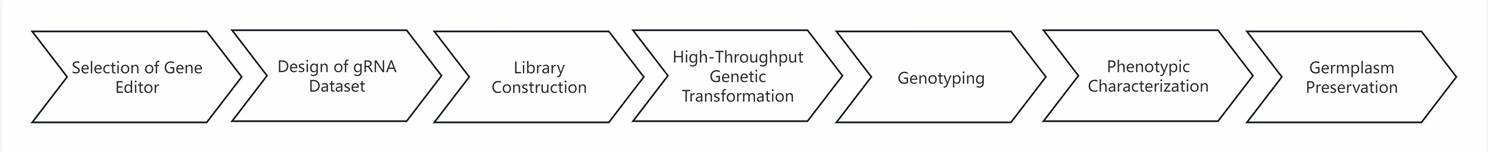 Figure 1 CRISPR Screening Process
Figure 1 CRISPR Screening Process
In the CRISPR screening process, the foremost task is the careful selection of an appropriate gene-editing tool. This section will focus on the applications and advantages of various editing tools, including CRISPR knock-out (CRISPRko), knock-in, base editors, prime editors, and CRISPR activation/interference (CRISPRa/i).
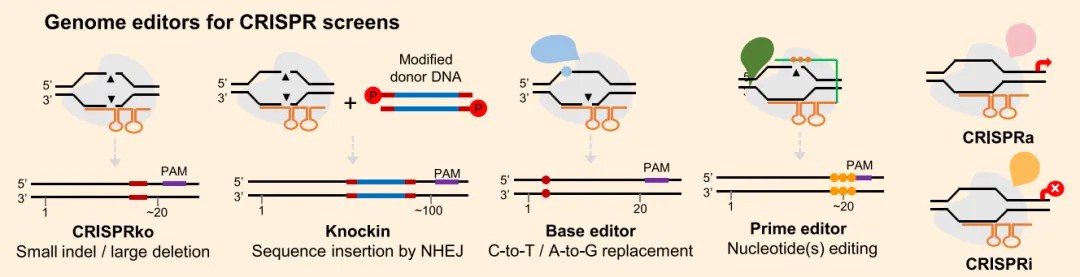 Figure 2: Selection of Appropriate Gene Editing Tools as the First Step in CRISPR Screening Process (Liu et al., 2023)
Figure 2: Selection of Appropriate Gene Editing Tools as the First Step in CRISPR Screening Process (Liu et al., 2023)
The CRISPR/Cas system, a modifiable nucleic acid endonuclease system, possesses the capacity for precise cleavage of target DNA. Its core components include the Cas protein, which encodes the nuclease, and the guide RNA (gRNA), which directs the system to the target sequence. In large-scale gene editing endeavors, the efficiency and stability of the editing tools are paramount. Numerous gene editing tools suitable for plant CRISPR screening have been identified.
Among the nucleases utilized in plant CRISPR screening, SpCas9 is one of the most frequently employed. The activity of SpCas9 is typically guided by a single guide RNA (sgRNA), which targets the 20 nucleotides upstream of a 5'-NGG-3' protospacer-adjacent motif (PAM) sequence. Under the direction of SpCas9, blunt-ended double-strand breaks (DSBs) in the DNA are induced, prompting nucleotide insertions or deletions (indels). These indels can lead to frameshift mutations within coding sequences, thereby effectuating the knockout of target genes (CRISPRko).
Although SpCas9-mediated gene knockout has become the predominant CRISPR screening tool, other gene editing tools exhibit substantial potential for broad applications. For instance, variants of SpCas9, such as Cas9 NG and SpRY, possess the ability to recognize a wider array of PAM sequences, thereby extending the potential applications of CRISPR editing. Cas9 NG is capable of recognizing various PAM sequences, including NG, NAC, NTG, NT, and NCG. In contrast, SpRY is largely unrestricted by PAM sequences, although it exhibits a preference for NNR and NRN PAMs.
Furthermore, the AT-rich PAM-recognizing Cpf1 is a significant gene editing tool. Due to the distal position of the Cpf1/Cas12a cleavage site relative to its PAM sequence, it typically generates larger deletions, such as those approximately 20 base pairs in length. Similarly, by fusing SpCas9 with specific enzymes, such as APOBEC3A and UNG, it is possible to induce comparable deletions. These tools exhibit unique advantages in screening cis-regulatory elements within promoters.
In the realm of mammalian cell research, the utilization of double-stranded oligodeoxynucleotides (dsODNs) that are phosphorylated at the 5' ends and contain phosphorothioate linkages at both termini has been shown to facilitate the precise integration of oligonucleotides into genomic sequences. This efficacy is primarily attributed to the enhanced stability conferred by the phosphorothioate modifications within the cellular environment and the promotion of non-homologous end joining (NHEJ) repair mechanisms by the 5' phosphorylation.
Base editors enable the editing of single nucleotides without inducing double-strand breaks (DSBs), representing a novel type of single-base substitution CRISPR screen. Several base editors have been developed, including cytosine base editors (CBEs), which convert C to T, and adenine base editors (ABEs), which convert A to G. Initial versions of these base editors exhibited insufficient editing efficiency for CRISPR screening applications.
Prime editors constitute another tool with significant potential for large-scale gene editing, enabling customized base editing. Despite relatively low initial editing efficiency, a series of improved tools have been developed, making prime editors promising for CRISPR screens. A study published in Nature Biotechnology, titled "An engineered prime editor with enhanced editing efficiency in plants," reported the development of an enhanced prime editor (ePPE). This editor, engineered by removing the RNase H domain from the reverse transcriptase and incorporating viral nucleocapsid proteins with nucleic acid chaperone activity, demonstrated improved efficiency at various endogenous loci, achieving an average 5.8-fold increase in editing efficiency without a significant rise in off-target effects. The ePPE was successfully used to generate herbicide-resistant rice plants with editing frequencies of 11.3%, compared to 2.1% with the original prime editor (Zong et al., 2022).
Beyond conventional gene editing, researchers have developed CRISPR activation (CRISPRa) and CRISPR interference (CRISPRi) systems by fusing dead Cas9 (dCas9) with transcriptional activators (e.g., VP64 and VPR) or repressors (e.g., KRAB and SRDX). These systems enable the upregulation or downregulation of target gene transcription, respectively (Tang et al., 2017; Pan et al., 2021; Zhang et al., 2022). CRISPRa and CRISPRi hold promise for application in plant CRISPR screens.
Bioinformatics analyses of candidate editing sites, such as quantitative trait loci (QTL) and genome-wide association studies (GWAS), are invaluable for CRISPR screens. Narrowing down candidate sites can significantly reduce the experimental burden. Following the selection of an appropriate editor, the next step is to design the gRNA library. It is essential to consider not only editing efficiency but also off-target effects. Numerous gRNA design websites are currently available (Table 1), with some programs enabling multiple target designs across the genome, such as CRISPy web and CRISPR Library Designer (available at https://github.com/boutroslab/cld) (Blin et al., 2016; Heigwer et al., 2016). These tools are particularly useful for obtaining comprehensive gRNA datasets that cover the candidate regions.
Table 1: gRNA Design Websites (Liu et al., 2023)
| Applicatior | Design programs | Description | Website |
|---|---|---|---|
| CRISPRko | CRISPy-web | CRISPy-web:An online resource to design sgRNAs for CRISPR applications |
https://crispy.secondarymetabolites.org/#/input |
| CRISPR-P | CRISPR-P 2.0:An Improved CRISPR-Cas9 Tool for Genome Editing in Plants |
http://crispr hzau.edu.cn/CRISPR2/ | |
| CRISPOR | CRISPOR:intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens |
http://crispor.org | |
| CHOPCHOP | CHOPCHOP v3:expanding the CRISPR web toolbox beyond genome editing |
https://chopchop.cbu.uib.no | |
| Base editing | PnB Designer | PnB Designer:a web application to design prime and base editor guide RNAs for animals and plants |
https://fgcz-shiny.uzh.ch/PnBDesigner/ |
| Betarge | BEtarget:A versatile web-based tool to design guide RNAs for base editing in plants |
https://skl.scau.edu.cn/betarget/ | |
| RGEN BE-Designer | Web-based design and analysis tools for CRISPR base editing |
http://www.rgenome.net/be-designer/ | |
| Prime editing | PlantPegDesigner | High-efficiency prime editing with optimized,paired pegRNAs in plants |
http://www.plantgenomeediting.net/ |
| PrimeDesign | PrimeDesign software for rapid and simplified design of prime editing guide RNAs |
https://primedesign.pinellolab.partners.org/ | |
| pegFinder | A web tool for the design of prime-editing guide RNAs | http://pegfinder.sidichenlab.org/ |
 Figure 3 illustrates that the size of a CRISPR screen is determined by the number of target genes, which can be effectively reduced through appropriate bioinformatics analysis. (Liu et al., 2023)
Figure 3 illustrates that the size of a CRISPR screen is determined by the number of target genes, which can be effectively reduced through appropriate bioinformatics analysis. (Liu et al., 2023)
Upon the completion of gRNA dataset design, a critical subsequent step involves the efficient cloning of single-guide RNAs (sgRNAs) into CRISPR/Cas plasmids. To ensure accuracy and even distribution of gRNAs within the plasmid library, it is advisable to incorporate a negative selection marker, such as ccdB, which significantly enhances cloning precision. Furthermore, to maintain a broad representation of gRNAs, it is essential to utilize a library with high coverage, typically exceeding 30× coverage.
Following the cloning process, thorough evaluation via next-generation sequencing (NGS) is necessary to verify the reliability and accuracy of the cloned constructs. Only after confirming the integrity and distribution of the gRNA clones through NGS can subsequent transformation procedures be initiated.
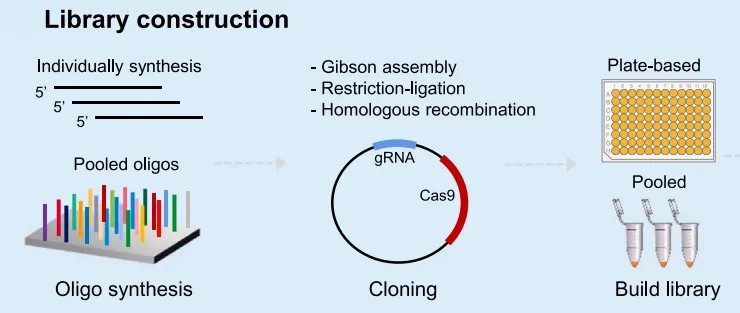 Figure 4 illustrates the synthesis of gRNA oligomers on a chip followed by cloning into suitable plasmid vectors to generate large-scale genome editing libraries (Liu et al., 2023).
Figure 4 illustrates the synthesis of gRNA oligomers on a chip followed by cloning into suitable plasmid vectors to generate large-scale genome editing libraries (Liu et al., 2023).
In mammalian CRISPR screening, a common approach involves transfecting a mixed gRNA library into cultured cells using lentiviral vectors, significantly advancing whole-genome analysis. Similarly, in plant research, a comparable strategy akin to mammalian CRISPR screening employs mixed Agrobacterium tumefaciens infections to achieve similar objectives.
Key considerations in mammalian cell transfection include ensuring each cell receives only one gRNA molecule to avoid the phenomenon of multiplex editing induced by multiple sgRNAs. Agrobacterium operates similarly to lentiviruses, integrating multiple exogenous DNA fragments into the host genome. The issue of multiplex editing in mammalian cell transfection has been effectively addressed by precisely controlling the multiplicity of infection (MOI)—the number of virus particles per cell. Specifically, using lower MOI values (0.3-0.5) ensures that the majority of cells predominantly receive one virus particle, achieving a "one-to-one" transfection effect (Shalem et al., 2015). Similarly, in mixed transformation experiments with rice callus tissues, using low MOI values of Agrobacterium also achieves a comparable "one-to-one" transfection effect (Lu et al., 2017). However, it is noteworthy that optimal Agrobacterium concentrations may vary significantly between different plant species such as tomato and Arabidopsis. Therefore, prior to conducting large-scale mixed genetic transformation work, thorough optimization and validation of concentrations are essential.
Unlike direct detection in mammalian cells, CRISPR screening in plants typically relies on tissue culture techniques to obtain edited plant collections for subsequent detection and analysis. Thus, compared to mammalian cells, implementing CRISPR screening in plants presents significantly increased challenges.
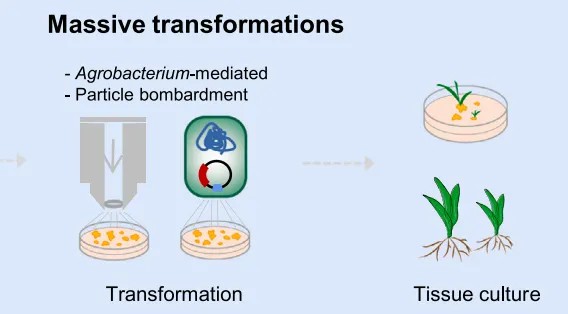 Figure 5 depicts the process of large-scale genetic transformation (Liu et al., 2023).
Figure 5 depicts the process of large-scale genetic transformation (Liu et al., 2023).
Following large-scale transformation, researchers obtain numerous edited plant samples that require extensive genotypic characterization. Next-generation sequencing (NGS) has become the mainstream method, with barcode technologies facilitating the efficient amplification and batch identification of gRNAs from mutants. Additionally, the Hi-TOM technique offers a highly efficient NGS-based method, requiring only two conventional PCR steps to swiftly construct high-throughput sequencing libraries. Complemented by the Hi-TOM online tool (http://www.hi-tom.net/hi-tom/), this approach automatically analyzes high-throughput sequencing data, effectively detecting various complex sequence information, including SNP molecular markers and gene editing mutations, to obtain detailed genotype data.
Upon completing genotypic analysis, the correlation between genotype and phenotype becomes more intuitive. However, due to potential off-target editing effects, further validation is necessary to ensure result accuracy. Unlike transient cell-based screening strategies in mammals, edited plants have the ability to produce seeds, enabling long-term preservation. Therefore, meticulously managing mutant libraries obtained through CRISPR screening technologies can accumulate valuable genetic resources for future scientific research and agricultural applications.
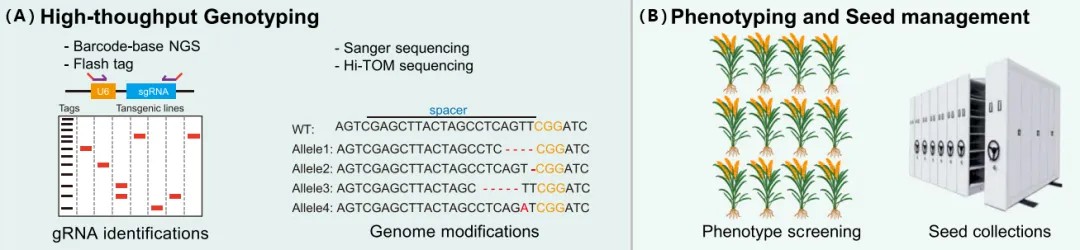 Figure 6 illustrates the process of high-throughput genotyping and phenotypic selection to manage seeds with desirable traits (Liu et al., 2023).
Figure 6 illustrates the process of high-throughput genotyping and phenotypic selection to manage seeds with desirable traits (Liu et al., 2023).
CRISPR screening in plants is primarily utilized for large-scale CRISPR knockout (CRISPRko) to construct whole-genome mutant libraries or analyze specific gene families. It facilitates promoter analysis by systematically deleting multiple genes to uncover novel traits.
Table 2: Summary of Plant CRISPR Screening (Liu et al., 2023)
| Application | Species | Number of | Genome editor | Description | |
|---|---|---|---|---|---|
| Gene | gRNA | ||||
| Large-scale CRISPRko |
Rice | 34234 | 88541 | SpCas9 | Genome-wide targeted knockout of all protein-coding genes in rice to obtain a mutant library |
| Rice | 12802 | 25604 | SpCas9 | Large-scale knockout of genes highly expressed in shoot base of rice | |
| Maize | 1244 | 1368 | SpCas9 | A knowledge-driven CRISPRko screen of candidate genes derived from GWAS in maize | |
| Rice | 1072 | 1166 | FLASH | Large-scale knockout of RLK genes in rice usingMultiplex mutagenesis of soybean genes involved in nodulation using an arrayed CRISPR library |
|
| Soybean | 102 | 70 | SpCas9 | Multiplex mutagenesis of soybean genes involved in nodulation using a pooled library | |
| Tomato | 54 | 162 | SpCas9 | Targeted knockout of an immunity-associated LRR-RLK subfamily in tomato using a pooled library |
|
| Promoter editing | Maize | 2 | 9;9 | SpCas9 | Editing of CLE genes to make weak promoter alleles to optimize ear size of maize that resulted in increased grain yield. |
| Rice | 1 | 39 | SpCas9 | Tiling deletion of IP41 promoter in ricegrain yield by decoupling panicle number and size | |
| Rice | 7;9 | SpCas9 | Quantitative regulation of Wx expression by promoter and 5'UTR-intron editing,resulted in improved grain quality |
||
| Tomato | 3 | 20;16;8 | SpCas9 | Cis-regulatory editing of SIWOX9,SICL V3,and SIWUS for improvement of productivity traits in tomato. |
|
| Orange | 1 | 5 | SpCas9 | Targeted modification of the CsLOBI promoter to enhance canker resistance in citnus | |
References
CD Genomics is transforming biomedical potential into precision insights through seamless sequencing and advanced bioinformatics.


