
We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
During the long process of evolution, each individual has developed unique genetic traits due to a variety of factors such as geography and environment. However, the genome of a single individual can no longer encompass all the genetic information of an entire species. As the cost of genome sequencing decreases, pan-genome research has risen rapidly in recent years, offering new possibilities to explore the genetic diversity and evolution of species.
Pan-genome research is a popular research direction. By sequencing (NGS and long-read sequencing) and assembling the genomes of different species and integrating the annotated gene sequences, we are able to gain comprehensive access to the genetic information of a species, and then analyze in-depth the genetic variation among individuals.
Please refer to our de novo sequencing and genome resequencing services for more information.
Our pan-genome service utilizes high-throughput sequencing and bioinformatics analysis to sequence and pan-assemble materials of different subspecies or individuals, thereby constructing a pan-genome map that enriches the genetic information of the species. The service can not only improve the gene set of the species, but also obtain the DNA sequence and functional gene information of specific populations or even individuals, providing a solid foundation for phylogenetic analysis and functional biology research.
In the field of plants, higher plants exhibit extremely high intraspecific genetic diversity to adapt to different growth environments. By constructing species pan-genomes, we can reconstruct the phylogenetic relationships between cultivated and wild varieties and study population-level recombination and cascade effects. Combined with the distribution of variants in each species in the pangenome, the expression of variants is detected using transcriptomic analysis, and trait-related variants are identified in combination with phenotypic information. With the variant information obtained through pan-genomic research, GWAS analysis is performed based on the structural variants to identify the loci associated with agronomic traits and provide genetic resource information for molecular breeding.
Compared with plants, the scope of animal pan-genomic research is more limited, mainly focusing on humans and domesticated animals. By constructing large-scale population genomes and focusing on species with certain common characteristics, it is possible to explore in depth the characteristic differences in the genomic structure of species, their formation history, convergent evolution, group characteristics, genome evolution and potential functions. By constructing pan-genomes of different strains, subspecies and variants, we can establish a comparison between core and non-core genomes, and compare the genotypic differences between groups in different geographical regions, thus revealing the environmental adaptations of species.
| Assembly | Type of assembly | Sequencing |
|---|---|---|
| de novo or resequencing | Fine assembly | HiFi(>30X) + Hi-C(>100X) + Illumina(>50x) |
| Non-fine assembly | HiFi(>30X) + Illumina(>50x) |
The short-read sequencing technique, while highly accurate, is limited by its read length of only about 100-200 nucleotides. Although it can reveal smaller variants like SNPs and inDels through contig-level assembly, it falls short in capturing larger variant types. Early pan-genome approaches often relied on mapping contigs to a reference genome, producing gene-centric pangenomes that may overlook complex structural variants crucial for gene regulation and genome evolution.
To overcome these limitations, we advocate for long-read sequencing, particularly leveraging PacBio and Nanopore platforms. By integrating next-generation and long-read technologies, this sequencing strategy constructs higher-quality genomes, enabling unbiased comparisons and revealing positional relationships and differences between them.
PacBio Single Molecule, Real-Time (SMRT) sequencing and Nanopore sequencing offer distinct advantages in genome assembly continuity and structural variation detection. These long-read technologies cover the entire genome without bias, providing accurate detection of various variants, including SNPs, indels, and structural variations. Their application holds significant promise in analyzing the genetic mechanisms underlying important crop traits. This, in turn, facilitates the design of genome-assisted breeding strategies, contributing substantially to the genetic improvement of crops.
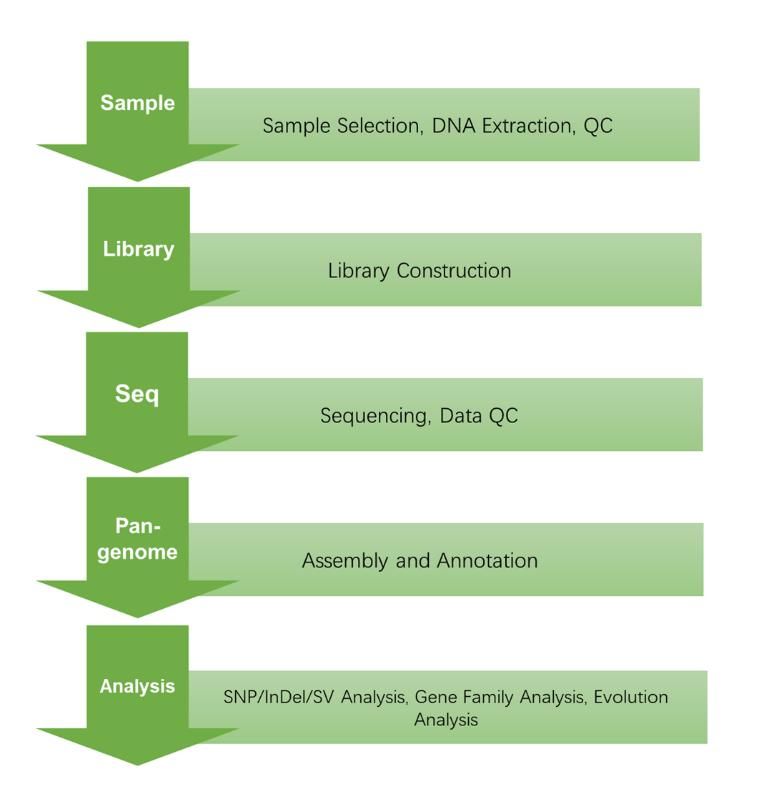 CD Genomics pan-genome service
CD Genomics pan-genome service
Construction of the Soybean Pan-Genome Atlas
Soybeans are a crucial source of edible oil and plant protein worldwide and a potential raw material for biofuels, holding a significant position in global agricultural trade. In comparison to traditional resequencing studies, pan-genome sequencing of multiple individuals provides a more comprehensive detection of genetic variations within the species. This approach offers a fresh perspective for in-depth understanding of the genetic characteristics of soybeans.
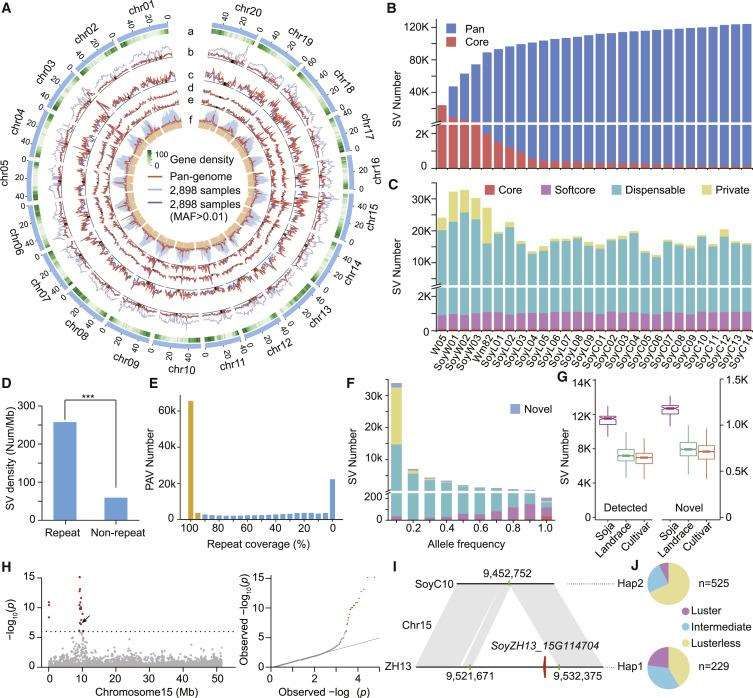 Genetic Variations from 29 Soybean Genomes and 2,898 Resequenced Accessions. (Liu et al., 2020)
Genetic Variations from 29 Soybean Genomes and 2,898 Resequenced Accessions. (Liu et al., 2020)
Reference
 Send a Message
Send a MessageFor any general inquiries, please fill out the form below.
CD Genomics is propelling the future of agriculture by employing cutting-edge sequencing and genotyping technologies to predict and enhance multiple complex polygenic traits within breeding populations.


