
 Sample Submission Guidelines
Sample Submission Guidelines
As an experienced provider of NGS services and a partner of Illumina, CD Genomics is committed to offering qualified metagenomic shotgun sequencing service. Shotgun metagenomic sequencing allows the taxonomic and biological functional characterization of polymicrobial communities in a cost-effective and time-efficient manner. We consistently deliver high-quality data with flexibility and bioinformatics analysis based on customers' requirements.
Introduction of Metagenomic Shotgun Sequencing
Microorganisms are present everywhere in nature. Studies reveal that microbes are critical to these environments and play an essential part in the ecosystem. It arouses attention for the interpretation of taxonomic composition of a complex microbial community. Shotgun sequencing has been proven as a fast sequencing strategy that uses random fragments. Metagenomic shotgun sequencing is a rapid and powerful tool for obtaining all genetic information in all organisms within a microbial community.
Metagenomic shotgun sequencing targets the entirety of the microbial genetic information contained in an environmental sample. The obtained community taxonomic profile can be further associated with the functional profile of known and unknown organism lineages. The complete sequences of protein-coding genes and full operons in the sequenced genomes can offer invaluable functional knowledge about the microbial communities inhabiting practical ecosystems under study. Metagenomic shotgun sequencing may provide genetic information on potentially novel biocatalysts or enzymes, genomic linkages between function and phylogeny for uncultured organisms, evolutionary profiles of community function and composition and much more. 16S/18S/ITS Amplicon Sequencing is another alternative for metagenomics studies. But this method only interprets the microbial biodiversity, without insights into function.
Advantages of Metagenomic Shotgun Sequencing
- Cultivation-independent
- Cost-effective, and time-efficient
- Provide comprehensive information on community biodiversity and function
- A wide range of applications in countless fields including the food and medical industries, environmental and disease studies, as well as biomarker research.
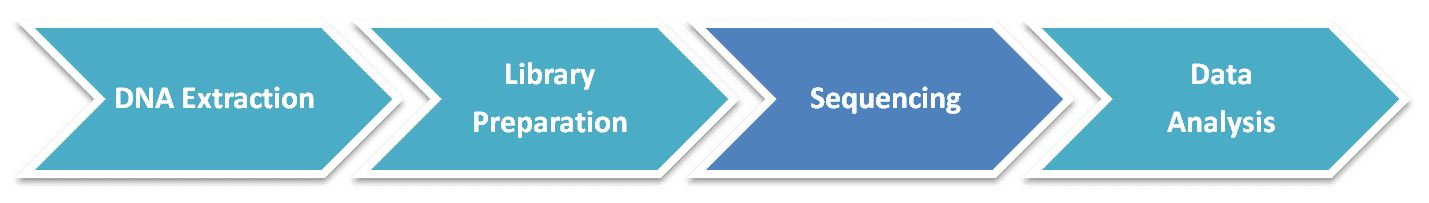
Metagenomic Shotgun Sequencing Workflow
Our highly experienced expert team executes quality management following every procedure to ensure comprehensive and accurate results. The general workflow for metagenomic shotgun sequencing is outlined below. Briefly, the sequencing library is constructed by DNA fragmentation, then the sheared DNA fragments are sequenced with Illumina HiSeq PE150 strategy. The raw data is processed by removal of adaptors, host genome sequence, and low-quality data.
Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
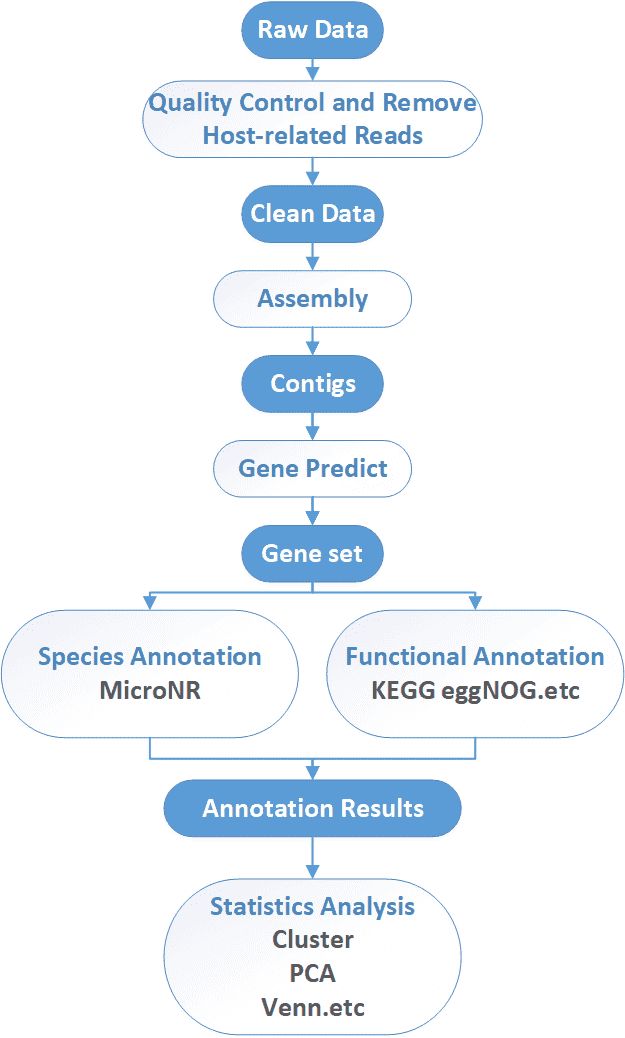
Bioinformatics Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Metagenomic Shotgun Sequencing for your writing (customization)
At CD Genomics, our experienced specialists will work closely with you to help to determine the optimal sequencing strategy based on specific research questions. With the state-of-the-art sequencing platforms and years of industry experience, we guarantee you high-quality data and integrated bioinformatics analyses. If you have additional requirements or questions, do not hesitate to contact us, our specialists would like to solve your problems.
Partial results are shown below:

Per base sequence conten.
Per sequence GC content.

Merged_abundance.

Abundance heatmap of family level.

KEGG_classification.

CAZy function classification.

Boxplot analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

PCoA analysis based on bray Curtis (A), binary jaccard (B), unweighted unifrac (C), and weighted unifrac (D).

UPGMA clustering tree.
1. How to prepare soil and faeces samples?
Soil samples: collect soil samples from 5-20 cm layer of soil, remove visible roots, and sieve the soil with 2 mm mesh. Mix up three sieved soil samples from three sites as one sample. Store it in a sterilized tube and place the tube below -20℃.
Faeces samples: store the faeces samples below -80℃. Empirically, fresh faeces contribute to isolating preferable DNA.
2. How to avoid host DNA contamination?
Host DNA contamination may affect the following bioinformatics analyses, especially when there's no reference genome for the host. Therefore, there are several matters needing attention when sampling. Try not to get close to tissues. Use relevant kit for DNA isolation. If there is reference genome for the host, remove the host DNA contamination by sequence alignment analysis.
3. What about 16S amplicon sequencing?
The 16S amplicon sequencing is an alternative choice for metagenomics studies since it reveals the microbial diversity and abundance from a range of environments, including soil, springs, ocean, roots, and the human gut. While powerful, amplicon sequencing has its limitations. First, the 16S amplicon sequencing would generate various biases associated with PCR, further leading to the widely varying estimates of diversity. Second, sequencing errors and incorrect assemblies can produce artificial sequences, i.e., chimeras. Third, 16S amplicon only provides insights into the taxonomic composition of the polymicrobial community, while can not illustrate the biological functions.
Reference
- Sharpton T J. An introduction to the analysis of shotgun metagenomic data. Frontiers in plant science, 2014, 5.
Shotgun metagenomic sequencing reveals freshwater beach sands as reservoir of bacterial pathogens
Journal: Water Research
Impact factor: 6.942
Published: 15 May 2017
Authors: Mahi M. Mohiuddin, et al., affiliated to the Department of Biology, McMaster University, Hamilton, ON, Canada
Backgrounds
Recreational waters may be heavily affected by adjacent sands, of which the bacterial concentrations can be 10-100 fold higher than those of the corresponding waters. Most previous studies have employed 16S amplicon sequencing with limited resolution at the species level and the inability to predict biological functions. It suggests that there is a need for a more comprehensive characterization of these recreational waters.
Materials & Methods
Sampling
Water and sand samples
Four sampling sites: Lakeside Beach, Fifty Point Beach of Lake Ontario; Long Beach and Nickel Beach of Lake Erie
Sequencing
Metagenomics shotgun sequencing
200 bp paired-end
HiSeq 2000
Taxonomic and functional annotations using MEGAN and BLASTx
Pathogen detection using CLARK
Statistics analysis (Alpha and beta diversity analysis, differential abundance testing)
Results
1、Community complexity and diversity
The rarefaction analysis (Figure 1) suggests that the taxonomic enrichment in sand samples is higher than that in water samples. The Shannon diversity revealed that there is no significant difference in the diversity index between lakes but a big significant difference in sand samples.
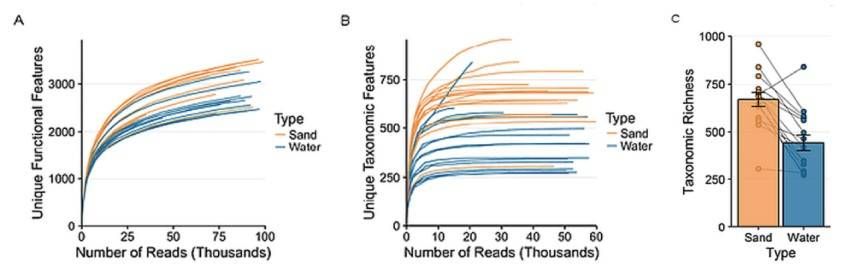 Figure 1. Rarefaction analysis was performed for both functional and taxonomic assignments. Functional assignments did not plateau and exhibited greater feature discovery as larger subsets were employed. Taxonomic assignment plateaued and sufficiently captured the bacterial diversity except one sample from waters of Long Beach of Lake Erie.
Figure 1. Rarefaction analysis was performed for both functional and taxonomic assignments. Functional assignments did not plateau and exhibited greater feature discovery as larger subsets were employed. Taxonomic assignment plateaued and sufficiently captured the bacterial diversity except one sample from waters of Long Beach of Lake Erie.
2、Taxonomic composition
There are no significant differences in the superkingdom composition between the sand and water samples. But the taxonomic composition at the phylum level showed significant differences (Figure 2). Proteobacteria were the most predominant group at most sites in the sand samples. Other predominant phyla are Bacteroidetes, Cyanobacteria, Verrucomicrobia, Firmicutes, and Planctomycetes. Additional, there were some unclassified phyla that were not observed in the water samples. All the sand samples contain taxa from a much wider array of phyla than the corresponding water environments (Table 1).
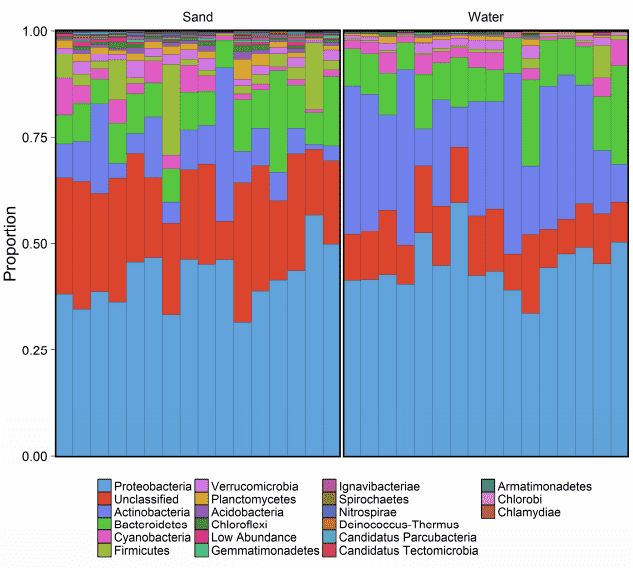 Figure 2. taxonomic composition at the phylum level.
Figure 2. taxonomic composition at the phylum level.
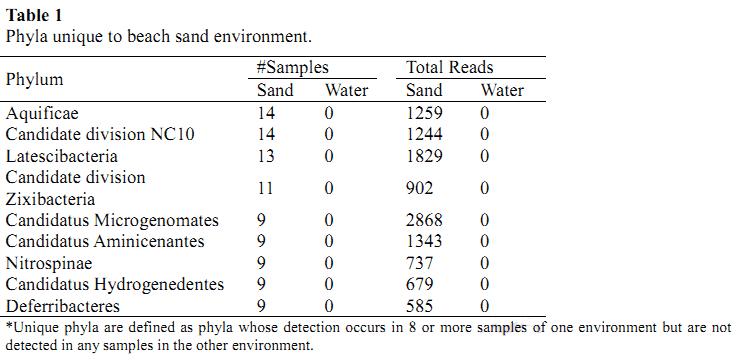
3、Functions
Interestingly, the differences in phylum composition did not have as large an effect on the functional capacity as determined by the SEED subsystem classifications. Functions affiliated to macromolecular processing and metabolism were detected with the greatest abundance across all samples. But some differences were seen in the capacity for more specific functions. One disparity between the two beach environments is the enrichment of functions pertaining to sulfur reduction in sand and sulfur oxidation in water, suggesting a dichotomy in sulfur utilization between the two communities. Other differences were also detected in the genetic capacity for spore formation, nitrosative stress, and hydrogenases, among other functions.
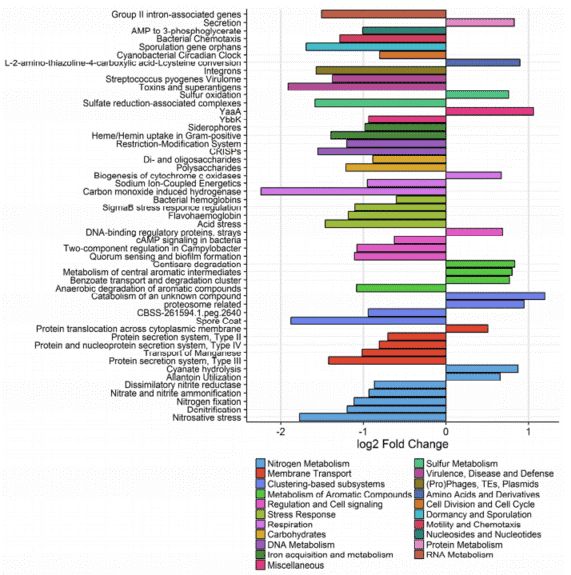 Figure 3. functions exhibiting differential abundance between beach environments.
Figure 3. functions exhibiting differential abundance between beach environments.
4、Potential human pathogens and fecal indicators in freshwater beaches
A total of 34 pathogens and fecal indicator were detected in both water and beach sands. The most abundant species was E. coli. Pseudomonas mendocina and Pseudomonas aeruginosa were also relatively abundant in both environments, and were significantly elevated in sand samples. Several low abundance pathogens were also detected from the Clostridium genus, with Clostridium botulinum exhibiting significant enrichment in water. Most sites examined had a presence of Vibrio spp. in the water but not in the sand.
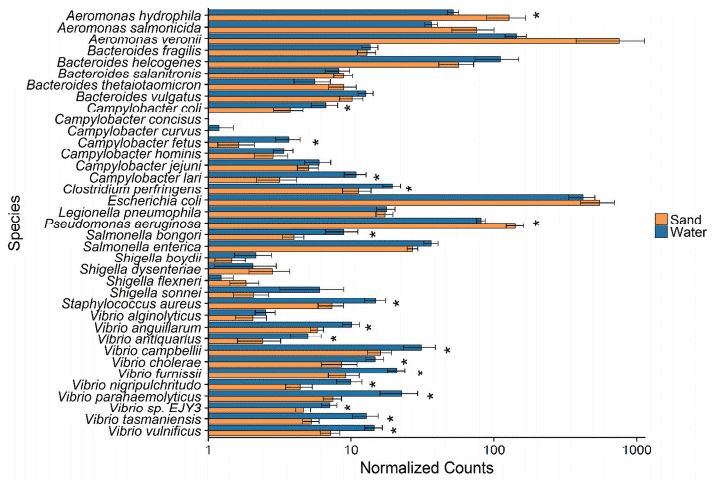 Figure 4. many pathogens exhibit differential abundance between beach environments.
Figure 4. many pathogens exhibit differential abundance between beach environments.
Conclusion
1. It suggests that shotgun metagenomic sequencing can potentially be employed to detect pathogens as evidenced by the detection of sequence homology to key pathogens, not easily detected by other traditional methods.
2. The data confirm that richness and diversity were significantly elevated in backshore sands compared to the corresponding waters.
3. Major differences were observed between the beach sand and water in terms of taxonomic composition at a broad level.
Reference
- Mohiuddin, M. et al. Shotgun metagenomic sequencing reveals freshwater beach sands as reservoir of bacterial pathogens, Water Research,2017, doi: 10.1016/j.watres.2017.02.057.
Here are some publications that have been successfully published using our services or other related services:
Hydrogen-Oxidizing Bacteria Are Abundant in Desert Soils and Strongly Stimulated by Hydration
Journal: mSystems
Year: 2020
Abundance and phylogenetic distribution of eight key enzymes of the phosphorus biogeochemical cycle in grassland soils
Journal: Environmental Microbiology
Year: 2023
Chitinase Chit62J4 Essential for Chitin Processing by Human Microbiome Bacterium Clostridium paraputrificum J4
Journal: Molecules
Year: 2021
Nutrient structure dynamics and microbial communities at the water–sediment interface in an extremely acidic lake in northern Patagonia
Journal: Frontiers in Microbiology
Year: 2024
Gene functions of the Ambystoma altamirani skin microbiome vary across space and time but potential antifungal genes are widespread and prevalent
Journal: Microbial Genomics
Year: 2024
Shifts in the rhizosphere microbiome and exudation profile of avocado (Persea americana Mill.) during infection by Phytophthora cinnamomi and in presence of a biocontrol bacterial strain
Journal: CABI Agriculture and Bioscience
Year: 2023
See more articles published by our clients.


