What is metatranscriptomic sequencing?
Metatranscriptomic sequencing provides direct access to culturable and non-culturable microbial transcriptome information by large-scale, high-throughput sequencing of transcripts from all microbial communities in specific environmental samples. Metatranscriptomic sequencing offers an opportunity to randomly sequence mRNAs as a unit for understanding the regulation of complex processes in microbial communities. The study of the metatranscriptome through Next-Generation Sequencing techniques allows us to obtain gene expression profiles from whole microscopical populations, providing new insights into poorly known biological systems and overcoming technical limitations related to individual bacteria isolation.
Challenges of metatranscriptomic sequencing
Although current metatranscriptomic techniques are promising, there are still several obstacles that limit their large-scale application. First, much of harvested RNA comes from ribosomal RNA (rRNA), and its dominating abundance can dramatically reduce the coverage of mRNA, which is the main focus of transcriptomic studies. To overcome this, some efforts have been made to effectively remove rRNA. Second, mRNA is notoriously unstable, compromising the integrity of the sample before sequencing. Third, differentiating between host and microbial RNA can be challenging, although commercial enrichment kits are available. This may also be done in silico if a reference genome is available for the host, as in the work of Perez-Losada et al. who considered the impact of host–pathogen interactions on the human airway microbiome. Finally, transcriptome reference databases are limited by their coverage.
Workflow of metatranscriptomic sequencing
The general workflow for metatranscriptomic sequencing is shown in Figure 1. To put it simply, the first step is to extract total RNA from the sample, and then to detect it. The qualified RNA is subjected to fragment screening, database construction and corresponding quality testing. The qualified library will be sequenced (mainly using Illumina sequencing platform). The raw data obtained by sequencing will be used for bioinformatics analysis.
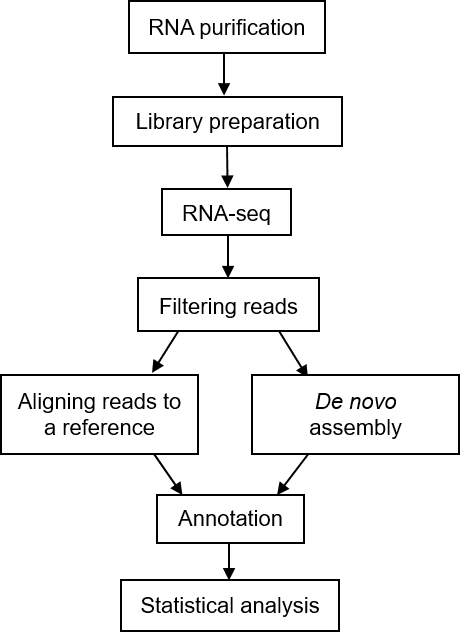
Figure 1. A typical workflow of metatranscriptomic sequencing (Peimbert M, et al. 2016)
The metatranscriptomics library preparation process is shown in figure 2. The two main strategies for mRNA enrichment are illustrated, either by using rRNA separation through means of hybridization with 16S and 23S rRNA probes, or by a depletion of rRNAs through means of a 5-exonuclease. Then, first strand of cDNA is synthesized by means of reverse transcriptase using random hexamers. And second strand of cDNA is synthesized by a DNA polymerase. Finally, sequencing adapters are attached to the cDNA strands, and this could be done either by PCR or by ligation.
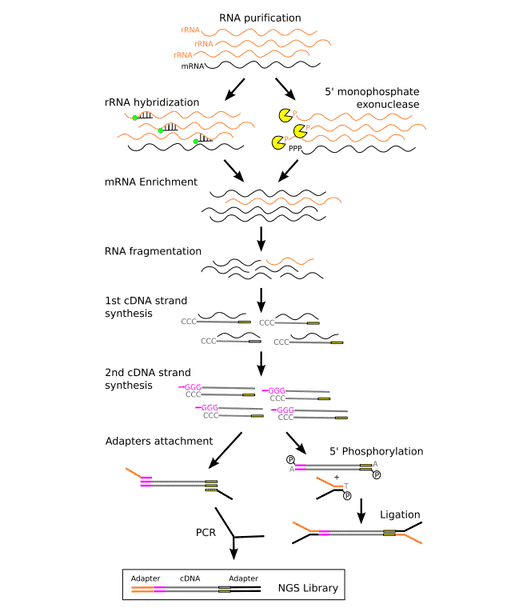
Figure 2. The metatranscriptomic sequencing library preparation process (Peimbert M, et al. 2016)
The overall process of metatranscriptomic sequencing bioinformatics is shown in Figure 3. The main steps are: filtering the readings, selecting the library between aligning the reference sequence and performing de novo assembly, annotation, statistical analysis, and uploading the original, assembled, and annotated data sets.
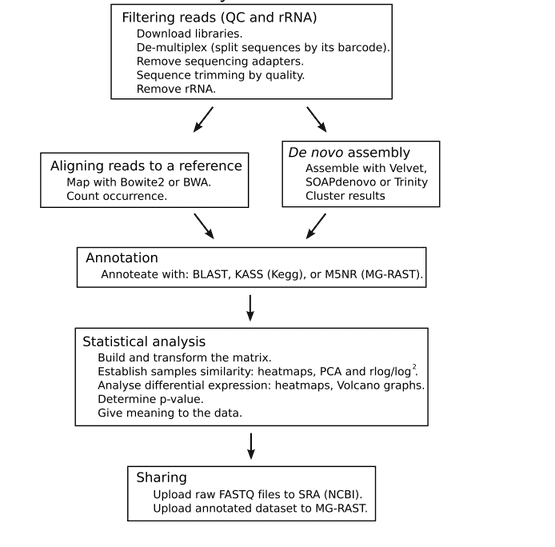
Figure 3. The metatranscriptomic sequencing bioinformatic overall process (Peimbert M, et al. 2016)
The applications of metatranscriptomics
Symbiotic bacteria (normal flora) play a key role in protecting us from pathogens, but under certain conditions they can overcome protective host responses and trigger pathological effects. Microbial population analysis can be used as an indicator of an individual's health status and as a powerful tool for the prevention, diagnosis and treatment of specific diseases.
- Assessment of microbiome–immune interactions
The effects of microbiota on the mucosal immune system are thought to be key to affect host physiology. A study of toll-like receptor 5 (TLR5) knockout (KO) mice is an interesting example of the use of metatranscriptomics to complement metagenomic characterization and 16S rRNA gene profiling of this microbial immune interaction. Metatranscriptomics analysis showed that flagellar motor-related gene expression was up-regulated in TLR5KO mice compared to wild-type mice. In this model, TLR 5 flagellin recognition causes the production of anti-flagellin antibodies, resulting in down-regulation of various bacterial flagellar motor genes, thereby inhibiting microflora. Deletion of TLR 5 results in reduced production of anti-flagellin antibodies, leading to upregulation of bacterial flagellar motor genes, thereby increasing the ability of bacteria in the gastrointestinal environment to disrupt the mucosal barrier.
- Studying microbiome small noncoding RNAs
The bacterial transcriptome includes small non-coding RNAs (sRNAs), which are typically between 50 and 500 bp in size and are involved in gene regulation. They regulate the translation or stability of the transcript by interacting with the 5'-untranslated region (UTR) of the target mRNA sequence. They are important for their ability to regulate important processes in bacteria, such as iron metabolism, virulence and quorum sensing, and to adapt quickly to changing environments. The emergence of next-generation sequencing methods has accelerated their identification of various bacteria, such as Salmonella and Bacillus subtilis. Next-generation sequencing methods also offer an opportunity to study bacterial sRNAs at the community level. For example, metatranscriptomics analysis of bacteria from different depths of the ocean suggests that sRNAs have a potential role in niche adaptation. Metatranscriptomics of the human activity gut microflora identified a number of sRNAs, although their role in gut microflora has not yet been elucidated.
Hundreds of drugs used today are derived from bacterial compounds. The study of metagenomes or metatranscriptomes in microbial communities offers new opportunities to explore innovative sources for drug discovery that are inaccessible today due to technical limitations in the isolation of these non-culturable microorganisms.
Microbial communities living on and around plants play a vital role in the nutrients needed for plant growth. In addition, the presence of specific micro-communities makes crops healthy and productive. Metagenomics and metatranscriptomics provide an opportunity to explore how microbial soil populations produce healthier and higher yielding crops.
Microorganisms are able to remove a wide variety of natural and synthetic harmful substances and convert them into other harmless compounds in humans and the environment. I don’t know how these microbial communities degrade harmful chemicals, but it provides new solutions for repairing and monitoring environmental pollution or improving drinking water purification methods.
Metagenomics and metatranscriptomics methods can be used to improve food quality, function and safety, and provide information related to metabolic activities of microbial communities.
At CD Genomics, we provide you with high-quality sequencing and integrated bioinformatics analysis for your metatranscriptomics project. If you have additional requirements or questions, please feel free to contact us.
References:
- Peimbert M, Alcaraz L D. A Hitchhiker’s Guide to Metatranscriptomic sequencing [M]// Field Guidelines for Genetic Experimental Designs in High-Throughput Sequencing. Springer International Publishing, 2016.
- Vanessa A P, Huang W, Victoria S U, et al. Metagenomics, Metatranscriptomic sequencing, and Metabolomics Approaches for Microbiome Analysis: [J]. Evolutionary Bioinformatics Online, 2016, 12(Supple 1):5-16.
- Dick G. Metatranscriptomic sequencing [M]// Genomic Approaches in Earth and Environmental Sci Maurice C F, Haiser H J, Turnbaugh P J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome[J]. Cell, 2013, 152(1-2):39-50.
- Warnecke F, Hess M. A perspective: Metatranscriptomics as a tool for the discovery of novel biocatalysts[J]. Journal of Biotechnology, 2009, 142(1):91-95.
- Jorth P, Turner K H, Gumus P, et al. Metatranscriptomics of the Human Oral Microbiome during Health and Disease[J]. Mbio, 2014, 5(2): e01012.
- O’Malley M A. Metatranscriptomics[M]. Springer New York, 2013.
- Cao Y, Fanning S, Proos S, et al. A Review on the Applications of Next Generation Sequencing Technologies as Applied to Food-Related Microbiome Studies: [J]. Frontiers in Microbiology, 2017, 8:1829.
- Bashiardes S, Zilberman-Schapira G, Elinav E. Use of Metatranscriptomics in Microbiome Research[J]. Bioinformatics & Biology Insights, 2016, 10(10):19-25.
For research purposes only, not intended for clinical diagnosis, treatment, or individual health assessments.

 Sample Submission Guidelines
Sample Submission Guidelines


