
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

What is RAD-seq?
Nowadays, the application of reduced-representation sequencing technology in large-scale and high-throughput SNP genotyping is a hot area of research. Restriction enzymes are employed to fragment genomic DNA, then some characteristic fragments are selected and subjected to the next generation sequencing technology for identification of genetic markers within the genome in a high-throughput way. The most widely used reduced-representation sequencing technology, restriction site-associated DNA sequencing (RAD-seq), is developed by researchers from the University of Oregon. Compared with paired-end and mate-pair libraries, RAD-seq technology with pooling method can build up to 96 sequencing libraries at one time, which is quite convenient for experimental operation and cost-effective. More importantly, it does not rely on the information of reference genome.
RAD-seq technology is a high-throughput sequencing of specific restriction enzyme fragments. Millions of genetic markers of polymorphism can be obtained by sequencing once, and RAD-seq has been widely used in ecology, genetics, genomics and other research fields. According to the type and quantity of restriction endonuclease used, RAD-seq can be divided into original RAD-seq, 2b-RAD, ddRAD, ezRAD, GBS (genotyping by sequencing) and other methods.
Advantages of 2b-RAD over other RAD-seq
2b-RAD, is a kind of RAD-seq technology, which is conducted on the basis of uniform fragments generated by type IIB restriction endonuclease. 2b-RAD effectively overcomes some of the limitations of RAD-seq, such as fussy library building process and varied lengths of restricted DNA fragments. We reviewed the main advantages of 2b-RAD over other RAD sequencing technologies, which are listed as follows.
(1) More kinds and numbers of markers. The number of loci obtained by different RAD-seq technologies varies greatly. Overall, 2b-RAD can get more loci, and is more suitable to study the evolutionary relationship, population structure, gene flow and other related problems. In addition to SNP, 2b-RAD can also provide dominant markers and CNV information.
(2) Independent tags. 2b-RAD tags are independent, it can reduce data redundancy and increase information content. Other Rad-seq technologies are likely to detect a sequence on either side of a restriction site, and the information provided by adjacent fragments is completely linked, equivalent to the redundancy of data. The 2b-RAD technique provides more information because the contact point is on both sides, making it impossible for two tags to be completely adjacent.
(3) All library sequenced. For 2b-RAD, all the enzyme fragments cut by restriction enzyme are used for sequencing, ensuring that the restriction sites are not lost. It makes the ultra-high density coverage of the genome, and the information is more complete. In theory, the average distribution distance of 2b-RAD tags on the genome is 2kb. That is, more than 200,000 tags can be obtained in a genome of 1G size.
(4) High repeatability. The process of 2b-RAD is simple and has high technical repeatability. The result is highly consistent after one sample was sequenced twice. It uses the N+1 strategy, so each project will perform a technical repeat for one individual by default. That is, the same sample will repeat the construction of library sequencing once, promising that the tag recurrence rate of the same sample should be no less than 95% between two experiments, and the mark recurrence rate should be no less than 85%.
(5) Uniform depth of sequencing. 2b-RAD technique generates equal length enzyme cutting tags of 33-36bp, which are enriched for the downstream high-throughput sequencing reaction, and the genome-wide high-throughput SNP screening and typing analysis are achieved through bioinformatics analysis. The uniform depth of sequencing guarantees the reliability and accuracy of each tag.
Table 1. The advantages of 2b-RAD over other Rad-seq technologies.
| Original RAD-seq | GBS | ddRAD | 2b-RAD | |
| Process | The process involves fragment selection and multi-step DNA isolation and purification | No physical interruption, fragment size selection and end repair | The process involves fragment selection and multi-step DNA isolation and purification | The process is simple, no physical interruption, fragment size selection and end repair |
| Fragment selection | 300-1000bp | ≤300bp | ≤300bp | 33-36bp |
| Coverage of restriction sites | Part | Part | Part | Whole |
| Technical repeatability | Low | Low | Low | High |
| Label density control | Poor | Poor | General | Flexible and controllable |
| Tag sequencing depth | Very different | Very different | Very different | Uniform |
| Marker type | SNP | SNP | SNP | SNP and dominant markers |
| Analysis | Removal efficiency was poor for false positive of SNP caused by repeated sequences | Removal efficiency was poor for false positive of SNP caused by repeated sequences | Removal efficiency was poor for false positive of SNP caused by repeated sequences | New data typing algorithm (iML) can effectively remove false positive SNP caused by repeated sequences |
Workflow of 2b-RAD
The core of 2b-RAD is the application of type IIb restriction endonuclease (Figure 1). This type of endonuclease recognizes six bases of double-stranded DNA, and then the upstream and downstream of the recognition site will cut off the DNA, resulting in a 27bp tag sequence. The 3' end of the tag is highlighted with 3 bases, which are also completely random in composition. The adaptor suitable for high-throughput sequencing platform is ligated to both sides of the tag, after PCR amplification, it can be used for sequencing.
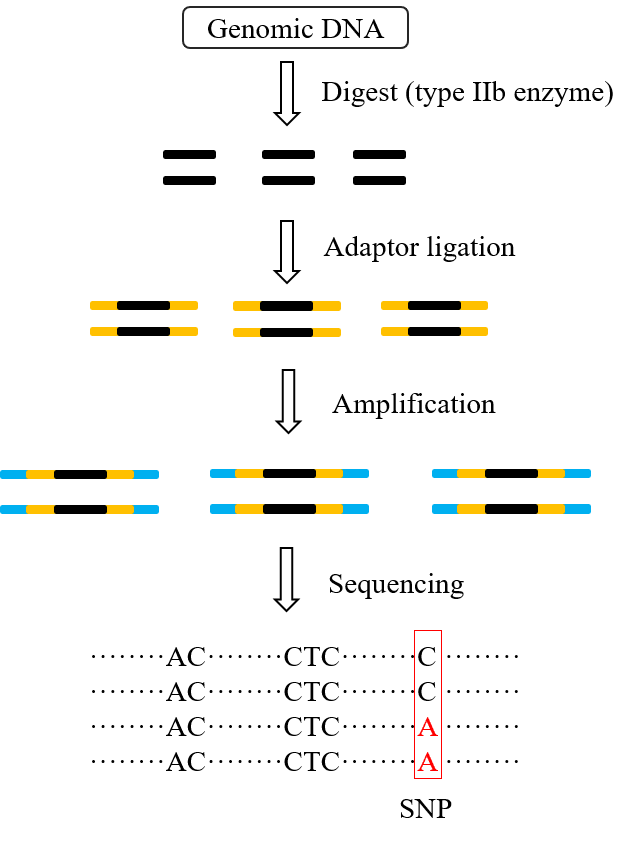
Figure 1. Preparation and sequencing of 2b-RAD tags.
(1) Sample preparation and digestion. Genomic DNA is extracted and its concentration, protein and RNA contamination are detected. The DNA is digested with restriction enzyme II. An additional sample can be digested simultaneously to detect the digestion efficiency by 1% agarose gel electrophoresis. The primary DNA band disappeared and became disperse, indicating a successful digestion.
(2) Adapter connecting. Specific adaptors are attached to the restriction fragments produced above. In this stage, the reduced tag representation (RTR) can be obtained to target a subset of the restriction sites for genotyping.
(3) Amplification. PCR amplification uses high-fidelity DNA polymerase and a set of primers. These primers can introduce sample-specific barcodes and the sequences required for bead enrichment on the sequencing system. An average of three amplification reactions are performed for each sample and merged. The merged PCR products were purified by electrophoresis.
(4) Sequencing. Qualified libraries are sequenced on either SOLiD or Illumina platforms. Standard BsaXI libraries for Illumina are serialized and then sequenced paired end by Hiseq X-ten, PE150 or Hiseq 2500 v2 platform.
(5) Quality control. SOLiD and Illumina platforms output reads of 35-bp length. The position of the terminal tag should be excluded in every time reading to eliminate the influence of artifacts. Then, long homo-polymer regions, excessive low-quality positions and unclear reads are eliminated. The remaining high-quality reads are used for the next analysis.
(6) Sequence alignment. For reference-based analysis, SHRiMP software package is used for alignment of high-quality reads against the reference database. For de novo analysis, CD-HIT software package is used for organisms that lack a completed genome sequence.
(7) Advanced analysis
I) High-precision linkage map construction. Based on the results of genotyping, Joinmap software can be used to build the genetic linkage map, and finally integrated the genetic map with MergeMap and other software.
II) Quantitative trait locus (QTL) localization. Based on the phenotypic data and the high-density linkage map, the precise QTL location of the quantity trait or quality trait can be achieved. Correlation analysis results can be used to assist in judging the reliability of QTL analysis results.
III) Population genetics. According to the number of tags between different groups and between different individuals within the group, the presence or absence of the same tag, the gene frequency and genotype frequency of SNP markers within the tag, population differentiation and evolution can be evaluated.
At CD Genomics, we provide you with high-quality sequencing and integrated bioinformatics analysis for your SNP genotyping project. If you have additional requirements or questions, please feel free to contact us.
References:


