
 Sample Submission Guidelines
Sample Submission Guidelines
CD Genomics provides single-cell DNA methylation analysis to understand the heterogeneity of genome-wide 5-methylcytosine (5mC) patterns.
The Introduction of Single-cell DNA Methylation Sequencing
DNA methylation is recognized as a principal contributor to the normal development and regulation of gene expression. It is essential for maintenance of cellular identity and is associated with a number of key processes including genomic imprinting, X-chromosome inactivation, repression of transposable elements, aging and carcinogenesis. The current method to research DNA methylation is only by bulk approach, such as whole genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS), and MeDIP sequencing. We combine the highly innovative single cell WGBS library preparation and Illumina next-generation sequencing technology to visualize genomic methylation states at single-cell resolution. Single cell whole genome bisulfite sequencing enables the discovery of cellular heterogeneity typically masked by standard, bulk methylation sequencing. We provide a streamlined workflow for making WGBS libraries. Input DNA is randomly fragmented during the initial bisulfite treatment step followed by WGBS library preparation. The procedure can accommodate ultra-low DNA input making it ideal for methylation analysis of precious, limited, and target-enriched samples.
What are the Advantages of Single-cell DNA Methylation Sequencing
- High-resolution single-cell methylation profiling: This methodological approach empowers the acquisition of intricate methylation landscapes at the single-cell level. It serves as a catalyst for delving into cellular heterogeneity and dynamic alterations, thereby propelling our comprehension of cellular diversity and transitions in state.
- Revealing cellular state transitions and evolutionary trajectories: Through the meticulous tracking of methylation dynamics in individual cells, this methodology unveils the intricate choreography of cellular state transitions and developmental trajectories. Such revelations offer invaluable glimpses into the molecular underpinnings of cellular development and evolution.
- Unearthing functional disparities among individual cells: Through meticulous comparative analysis of methylation profiles, this method illuminates functional disparities among individual cells, thereby elucidating potential mechanisms governing cellular function and epigenetic regulation.
- Minimal DNA input requirement: Characterized by its minimal DNA input demand, this technique facilitates the attainment of ultra-low detection thresholds with only a sparse number of cells.
- Bioinformatic scrutiny: The data garnered paves the way for exhaustive and intricate bioinformatic analyses, furnishing rich information content for meticulous examination.
- Assessment of data quality: Parameters such as mapping ratio and redundancy closely mirror those of conventional WGBS data, thereby ensuring the integrity and reliability of the data obtained.
What are the Application of Single-cell DNA Methylation Sequencing
In the realm of scientific inquiry, investigations into methylation patterns in single cells and minute, rare samples are predominantly applied across various research domains such as elucidating tumor etiology, cancer studies, pre-implantation embryo diagnostics, early embryonic development, germ cell recombination, stem cell research, and cellular heterogeneity exploration. These endeavors often involve samples including single cells, minute cell populations, and trace amounts of DNA. This methodology is particularly suited for analyzing elusive specimens that are difficult to obtain and tissue samples exhibiting significant cellular heterogeneity.
- Tumor heterogeneity: Unveiling the methylation landscape of cancer cells.
- Cell differentiation: Charting epigenetic regulatory maps of cellular differentiation.
- Immune microenvironment: Elucidating the epigenetic regulatory mechanisms underlying immune cell activation, differentiation, and other processes.
- Neuroscience: Subtype classification of neural cells and exploration of the molecular mechanisms underlying neuro-related disorders.
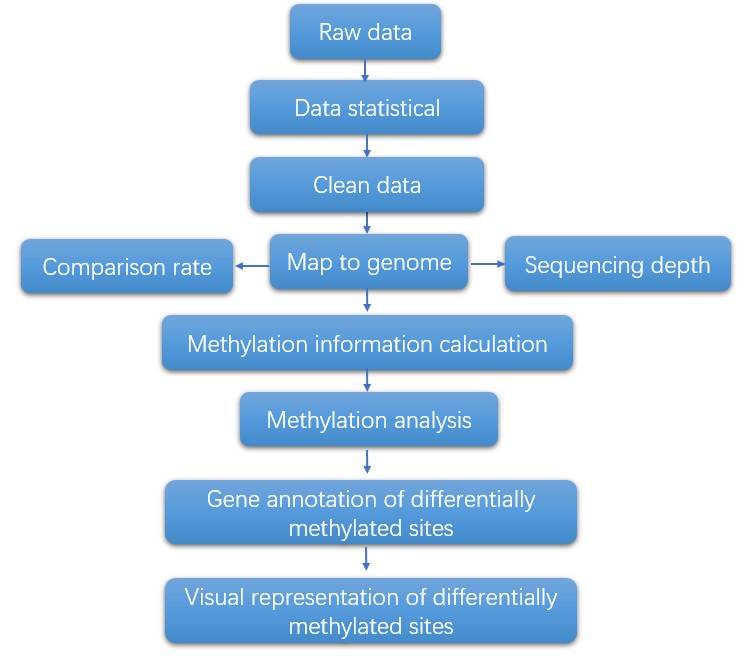
Single-cell DNA Methylation Sequencing Workflow
With post-bisulfite library preparation for WGBS, the workflow can be completed in a few hours. The general workflow for single-cell methylation sequencing is outlined below.

Service Specification
Sample Requirements
|
|
 |
Sequencing
|
 |
Bioinformatics Analysis
|
Analysis Pipeline
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Single-cell DNA Methylation Sequencing for your writing (customization)
CD Genomics's Single-Cell Sequencing conference focuses on the links between cell variation in tissues and organ function and further elucidates the origins of diseases. If you have additional requirements or questions, please feel free to contact us.
Reference:
- Gravina, S et al. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biology, 2016 17(1):150.
1. What are the advantages of single-cell DNA methylation sequencing?
The utilization of single-cell DNA methylation sequencing confers multiple merits. Primarily, it provides the means to scrutinize the heterogeneity observed among individual cells, trace uncommon cellular populations, and closely monitor dynamic alterations in DNA methylation during phases of development or progression of diseases. This elucidates cell-specific epigenetic fingerprints and facilitates the scrutiny of epigenetic regulation at an individual cellular level.
2. What are the challenges associated with single-cell DNA methylation sequencing?
Despite a plethora of benefits, the practice of single-cell DNA methylation sequencing is not devoid of challenges. Predominantly, it involves intricate technical nuances such as the isolation and amplification of individual cells, which, coupled with the financial burden and intricacy of sequencing singular cells, poses substantive obstacles. Furthermore, considerable challenges are encountered during data analysis given the sparsity of single-cell epigenomic data and the subsequent requirement for bespoke bioinformatics apparatus.
3. How is single-cell DNA methylation sequencing advancing our understanding of biology and disease?
Single-cell DNA methylation sequencing is trailblazing our understanding of variations between individual cells, their roles in biological developmental processes, and the mechanisms behind diseases. By bringing to light epigenetic nuances within individual cells, the technique is demystifying the intricacies of cell fate determination, tissue regeneration, and the trajectory of diseases. It possesses great potential in unveiling novel therapeutic objectives, thereby augmenting the diagnosis and treatment of an array of diseases, including malignancies and neurological disorders.
4. What are the differences between single-cell methods based on RRBS and single-cell methods based on WGBS?
Coverage:
RRBS: RRBS is specifically engineered to examine CpG island regions within the genome through a mechanism known as restriction enzyme digestion. Although its genomic breadth is delimited, it offers remarkable depth coverage owing to this specific targeting.
WGBS: Contrary to RRBS, WGBS surveys the complete genome's methylation terrain which encompasses CpG islands as well as chromatin regions characterized by weak methylation. Despite providing more expansive genomic coverage, the depth coverage is often compromised.
Depth and Resolution:
RRBS: Predominantly, due to its specialized enrichment of CpG island regions, RRBS frequently delivers robust depth coverage albeit the resolution is mitigated, which impairs the ability to discern non-CpG island regions present in the genome.
WGBS: Alternately, considering that WGBS sweeps across the complete genome, its depth may be potentially hampered, especially in single-cell sequencing contexts, when juxtaposed with RRBS. Regardless, the superior resolution offered by WGBS facilitates the capture of broader genomic methylation patterns.
Data Volume and Analytical Complexity:
RRBS: Given its constrained target spectrum, the data volume generated by RRBS is relatively compact which simplifies subsequent data processing.
WGBS: In contrast, WGBS typically results in tremendous data volume, thereby necessitating an augmented storage and computational capacity. Consequently, this often culminates in more complex data handling and interoperation procedures.
Applications:
RRBS: Where investigating methylation within CpG island domains is concerned, RRBS shines as an optimal choice. It is particularly proficient at delving into methylation incidents that are vital to the functionality of the genome and its epigenetic regulation.
WGBS: On the other hand, WGBS presents itself as a superior choice for conducting comprehensive elucidation of genome-wide methylation patterns. It is instrumental in disclosing global methylation contours and aids in studying methylation events that are specific to cell-types and those prone to dynamic shifts.
Single-cell DNA methylome sequencing of human preimplantation embryos
Journal: Nature genetics
Impact factor: 27.125
Published: 18 December 2017
Background
In mammalian genomes, cytosine, primarily in CpG dinucleotides, undergoes methylation under the catalysis of DNA methyltransferases. Research has revealed that DNA methylation is crucial for numerous biological processes, including gene expression repression, regulation of transposon transcriptional activity, X-chromosome inactivation, and maintenance of genomic imprinting. Previous studies have indicated a large-scale DNA demethylation during preimplantation embryonic development. However, current research data suggests that following fertilization by sperm and oocyte fusion, alongside massive DNA demethylation in early human embryos, there is also extensive highly-specific de novo DNA methylation. This implies that the 'net result' of global DNA demethylation during the first round of DNA methylome reprogramming in early human embryos actually manifests as a dynamic equilibrium produced by the contest of the extensive organized DNA demethylation process and localized DNA methylation.
Methods
- Human gametes
- Human preimplantation embryos
- Genomic DNA extraction
- Whole-genome bisulfite sequencing
- Sequencing library construction
- Sequencing read quality control and alignment
- DNA methylation level estimation
- SNP calling and parental genome tracing
- Allele-specific gene expression
- Differentially methylated regions
Results
In this investigation, we conducted single-cell PBAT DNA methylome sequencing analysis on a cohort comprising 480 individual cells. This cohort included 50 human oocytes, consisting of 42 mature oocytes and 8 germinal vesicle (GV) oocytes, along with 23 single sperm cells and 62 preimplantation embryos. The sequencing endeavor yielded a substantial dataset, amounting to 6.5 Terabytes. On average, each individual cell underwent sequencing for 8.4 Gigabytes, facilitating coverage of approximately 10.8 million CpG sites (≥1×). Notably, our analysis identified 3 aneuploid oocytes and 33 embryos harboring at least one aneuploid blastomere. These aberrant specimens were subsequently excluded from further analytical scrutiny.
Detailed investigation uncovered three waves of global demethylation during preimplantation embryo development. The first wave, occurring within the initial 10 to 12 hours post-fertilization, demonstrated a significant decrease in DNA methylation levels in both the paternal and maternal genomes. Subsequent waves of demethylation were observed from the late zygote to the two-cell stage and from the eight-cell to the morula stage, with distinct methylation dynamics in genomic regions enriched for enhancers, gene bodies, introns, and SINEs.
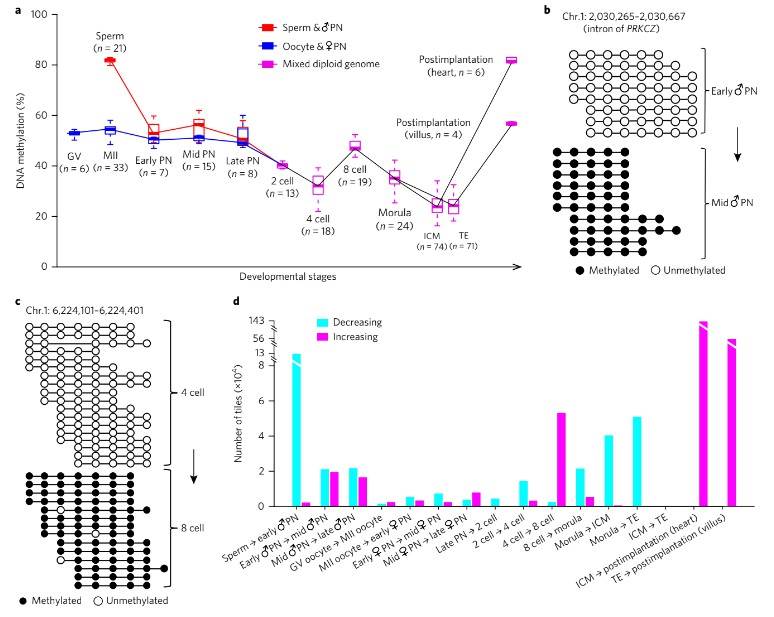 Fig. 1. De novo methylation patterns in early human embryos.
Fig. 1. De novo methylation patterns in early human embryos.
Additionally, drastic de novo DNA methylation events were observed during preimplantation development, particularly during two distinct waves: from the early male pronuclear to mid-pronuclear stage and from the four-cell to the eight-cell stage. These de novo methylated regions were predominantly enriched for repeat elements such as SINEs, LINEs, and LTRs, suggesting a mechanism for repressing their transcriptional activity.
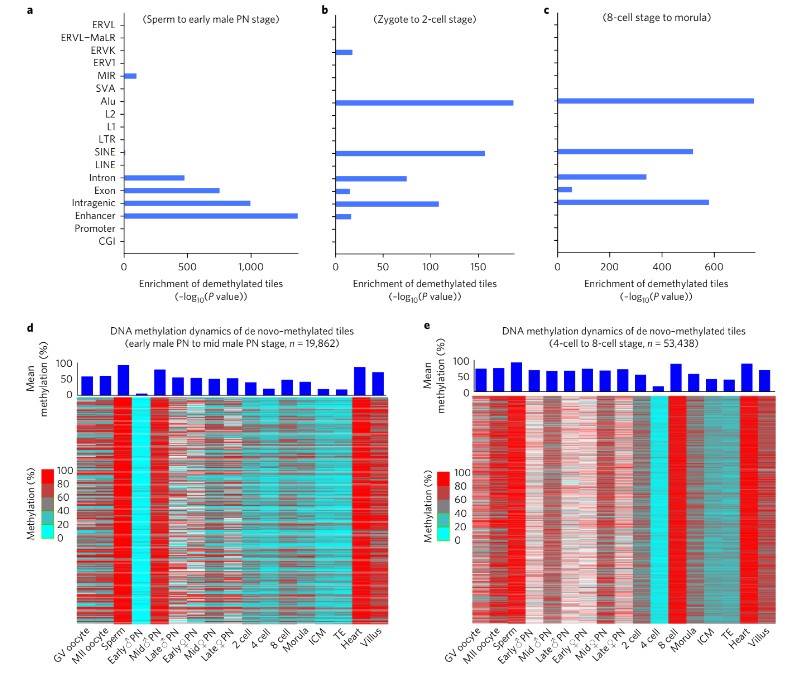 Fig. 2. DNA methylation dynamics of de novo–methylated regions and enrichment analysis of demethylation regions.
Fig. 2. DNA methylation dynamics of de novo–methylated regions and enrichment analysis of demethylation regions.
Furthermore, differences in demethylation dynamics between the paternal and maternal genomes were elucidated, with the paternal genome exhibiting faster demethylation rates than the maternal genome during early embryonic stages. Notably, the methylation pattern favoring the maternal genome persisted throughout preimplantation and postimplantation development across embryonic and extra-embryonic lineages.
Finally, examination of differentially methylated regions (DMRs) between oocytes and sperm unveiled specific genomic sites enriched for paternal and maternal DMRs. These findings underscore parental-specific DNA methylation patterns that play pivotal roles in modulating gene expression and genomic imprinting throughout embryonic development.
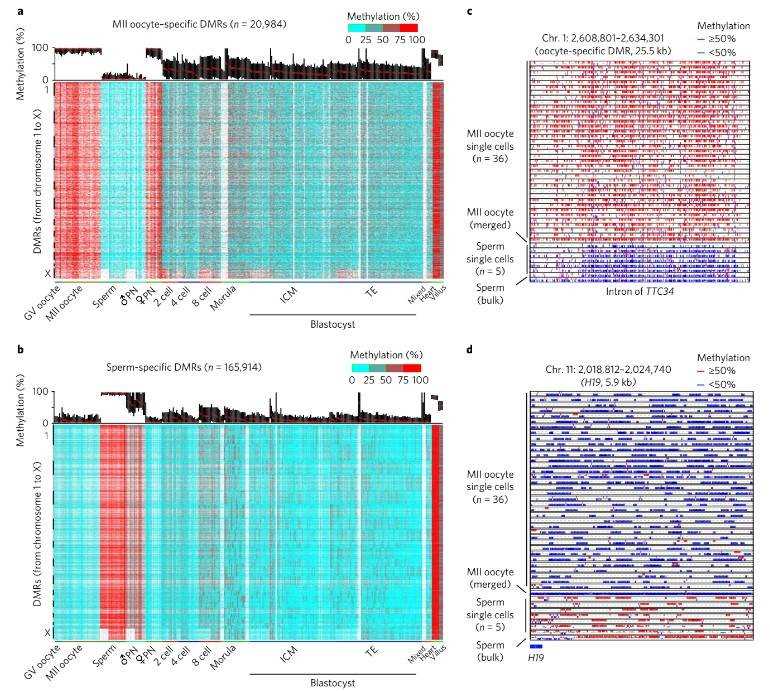 Fig. 3. Characterization of gamete-specific differentially methylated regions.
Fig. 3. Characterization of gamete-specific differentially methylated regions.
Conclusion
In this investigation, we embarked on a journey to delve deeper into the dynamic intricacies of DNA methylation reprogramming at the single-cell level. Leveraging cutting-edge high-throughput sequencing technology, we embarked on single-cell whole-genome DNA methylation analysis. This pioneering approach enabled us to systematically scrutinize pivotal stages of human pre-implantation embryonic development with unprecedented precision, unraveling insights at a single-cell, single-base resolution. Our explorations yielded several landmark discoveries:
(1) Unveiling specific de novo DNA methylation: For the first time, we unveiled the presence of a substantial volume of specific de novo DNA methylation throughout the course of human pre-implantation embryonic development.
(2) Revelations on parental genome methylation dynamics: In a groundbreaking revelation, we observed a reversal in residual methylation levels on parental genomes, commencing from the two-cell embryo stage. Intriguingly, within the same single cell, the residual methylation levels on the maternal genome significantly surpassed those on the paternal genome.
(3) Insights into asymmetrical DNA methylation distribution: Our study uncovered, for the first time, that the asymmetrical distribution of DNA methylation during early embryonic cleavage holds promise as a tool for tracing the genetic lineage of individual cells within the same embryo.
Reference:
- Zhu P, Guo H, Ren Y, et al. Single-cell DNA methylome sequencing of human preimplantation embryos. Nature genetics, 2018, 50(1): 12-19.


